

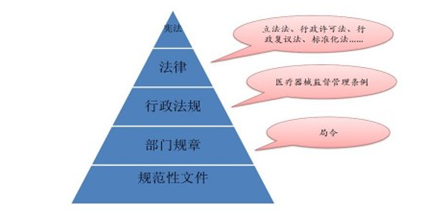
医疗器械注册法规,是各个国家、地区的政府和主管当局监管医疗器械的依据。由于医疗器械不同于其它普通商品,它关系到操作者、使用者或患者的安全健康问题,为此,全世界所有发达经济体和绝大多数发展中国家均建立了医疗器械监管体系,以保障公众身体健康和生命安全。目前各个国家、地区的医疗器械监管体系不尽相同,但通常都是类似于如下中国监管体系的金字塔式结构。
上图为金字塔式结构的中国医疗器械监管法规体系,就第二层的法律而言,目前中国尚未制订专门针对医疗器械监管的类似于《药品法》的法律;第三层是由国务院发布的《医疗器械监督管理条例》,通常包括上市前和上市后监管要求;第四层是由国家医疗器械监管部门发布的部门法规,指导医疗器械制造商在设计、生产、注册登记、销售、售后等各个环节符合法规要求。其他国家与中国医疗器械监管体系类似,但部分国家和地区医疗器械涵盖动物用医疗器械,本报告不涉及该部分。本报告的重点研究范畴是对应于中国监管体系第三、四层的医疗器械法规。下将针对美国、欧盟、日本和澳大利亚等国法律法规,及对全球医疗器械法规协调做出重大贡献的 GHTF、IMDRF、AHWP 组织做综述性介绍。
1. 各国医疗器械法规综述
1.1美国
美国食品药品管理局(Food and Drug Administration,FDA)是美国人类和健康服务部(Department of Health & Human Services,DHHS)的下设机构之一,负责对药品、食品、化妆品、医疗器械、兽药等产品进行全面监督管理,其负责医疗器械的部门是 CDRH(Center for Devices and Radiological Health)。作为一个在世界范围备受关注的监管机构,美国 FDA 经历了一百多年的发展历程,建立了相对完整的法律法规体系。
其医疗器械相关的主要法律有:
l Federal Food, Drug and Cosmetic Act
联邦食品药品化妆品法案
l Medical Device Amendments Act
医疗器械修订法案
Food and Drug Administration Modernization Act
食品药品监督管理局现代化法案
Medical Device User Fee And Modernization Act
医疗器械用户收费和现代化法案
医疗器械相关的主要法规有:
l Code of Federal Regulation (CFR)Title 21
联邦法规第 21 卷
指南性文件数量巨大,属于非强制性法规要求,举例如下:
l Guidance for industry: Part 11, Electronic Records; Electronic
Signatures — Scope and Application
工业指导文件:电子记录和电子签名范围和应用 Part 11
l Guidance for industry: Non-Clinical Engineering Tests and Recommended
Labeling for Intravascular Stents and Associated Delivery Systems
工业指导文件:心血管支架和相关的输送系统临床前的工程测试和推荐的标示
1.2欧盟
欧盟委员会(Commission of European Union)是欧盟的常设执行机构,且是欧盟唯一有权起草法令的机构,负责实施欧盟条约和欧盟理事会做出的决定,处理日常事务,代表欧盟对外联系和进行贸易等方面的谈判。目前,欧盟委员会已颁布实施三个医疗器械指令,以使医疗器械投放市场的规定 协调一致。
l Council Directive 93/42/EEC on Medical Devices(MDD)
医疗器械指令 93/42/EEC
l Council Directive 90/385/EEC on Active Implantable Medical Devices(AIMD)
有源植入医疗器械指令 90/385/EEC
l Council Directive 98/79/EEC on In Vitro Diagnostic Medical Devices(IVDD)
体外诊断医疗器械指令 98/79/EEC欧盟委员会也制订了以下医疗器械相关的主要法规,例如:
l Reclassification of hip, knee and shoulder joint replacements ,Commission (Directive 2005/50/EC)
臀部,膝盖和肩部连接假体的重新分类
l Reclassification of breast implants (Directive 2003/12/EC)
胸部植入物的重新分类
l Electronic instructions for use of medical devices (Commission
Regulation (EU) No 207/2012)
电子版医疗器械说明书
l MD manufactured utilising tissues of animal origin (Directive2003/32/EC)
采用动物来源的材料制造的医疗器械
Common Technical Specification on IVD
诊断试剂的常用技术要求
Eudamed - European Databank on Medical Devices Eudamed -欧盟医疗器械数据库
欧盟委员会也制订医疗器械相关的指南(超过 30 个),例如:
l Guidance: Classification of medical devices MEDDEV
2.4医疗器械分类的指南
l Clinical evaluation: Guide for manufacturers and notified bodies
MEDDEV 2.7/1针对制造商和公告机构在临床评估方面的指南
1.3日本
日本厚生劳动省(Ministry of Health, Labor and Welfare,MHLW)是日本负责医疗卫生和社会保障的主要部门。厚生劳动省于 1960 年首次颁布《药事法》(Pharmaceutical Affairs Law),其目的是为了确保药品包括诊断试剂、化妆品、医疗器械的品质,及其安全性和有效性。随后该法律在 2002 和 2003 做过修订,修订内容于 2005 年正式生效。
医疗器械相关法律
l Pharmaceutical Affairs Law (PAL)药事法
医疗器械相关主要法规,例如:
l Enforcement Ordinance for PAL
药事法施行令
l Enforcement Regulations for PAL
药事法施行规则
l GQP Ministerial Ordinance
良好质量管理省令
l QMS Ministerial Ordinance
质量管理体系省令
l GLP Ministerial Ordinance
良好实验室规范省令
l GCP Ministerial Ordinance
良好临床试验规范管理省令
l GVP Ministerial Ordinance
良好警戒规范省令医疗器械通知文件相当于指南性文件,种类和数量较多,举例如下:
l MHLW No.298:Classification of Medical Devices 医疗器械分类的通知文件
l MHLW No.2:Sterilization residual limitation 灭菌残留的限度的通知文件
1.4澳大利亚
治疗品管理局(Therapeutic Goods Administration,TGA)是澳大利亚政府健康和老龄部所属的一个监管机构,负责监督管理澳大利亚的治疗品(包括药品、医疗器械、基因科技和血液制品)。TGA 开展一系列的评审和监督管理工作,以确保在澳大利亚提供的治疗品符合适用的标准,并保证澳大利亚社会的治疗水平在一个较短的时间内达到较高的水平。
医疗器械相关法律:
l Therapeutic Goods Act
治疗品法案
l Therapeutic Goods (Charges) Act
治疗品(收费)法案
医疗器械相关法规:
l Therapeutic Goods Regulations
治疗品法规
l Therapeutic Goods (Medical Devices) Regulations
治疗品(医疗器械)法规
l Therapeutic Goods (Charges) Regulations
治疗品(收费)法规
医疗器械相关指南文件
l Australian regulatory guidelines for medical devices (ARGMD)
澳大利亚医疗器械法规指南
1.5 全球协调工作组 Global Harmonization Task Force(GHTF)全球协调工作组(Global Harmonization Task Force,GHTF)是全球最重要的国际医疗器械法规协调的组织,成立于 1992 年。它是一个为应对日益增长的医疗器械法规的全球统一协调的需求而成立的志愿组织。其五个发起成员为美国、欧盟、日本、加拿大和澳大利亚,主席由发起国的法规主管机构轮流担任。自 2006 年起,会员也增加 AHWP、ISO 及 IEC 等 3 个组织。GHTF 工作目的是在于建立医疗器械的安全性、有效性评估与品质系统检查等国际技术规范,协助各个国家医疗器械监管的主管当局建立技术共识,采用相同的要求执行医疗器械管理,奠定国际间互相承认的基础,以推动国际贸易。同时 GHTF 也是作为信息交换平台,分享会员的医疗器械监管的经验。GHTF 主要的途径是通过出版和发布被成员国政府认可的基本的监管指导性文件,和相对应法规的统一司法解释指南,给各国法规主管机构提供监管参考。GHTF 组织在 2012 年 11 月正式宣布关闭,被国际医疗器械监管者论坛(TheInternational Medical Device Regulators Forum,IMDRF)取代。
1.6 国际医疗器械监管者论坛 The International Medical Device Regulators Forum(IMDRF)国际医疗器械监管者论坛(The International Medical Device Regulators Forum,IMDRF)成立于 2011 年 10 月,由来自澳大利亚、巴西、加拿大、中国、欧盟、日本、美国和世界卫生组织(WHO)医疗器械监管机构的代表在渥太华召开会议,宣布成立国际医疗器械监管者论坛。IMDRF 将以论坛的形式讨论未来医疗器械监管协调的方向。IMDRF 是一个在医疗器械全球协调工作组(GHTF)的工作基础上,由来自全球的医疗器械监管者自愿成立的组织,该组织旨在加速国际医疗器械监管的协调和融合。
目前 IMDRF 管理委员会的成员国和 GHTF 基本相同,有美国、欧盟、日本、加拿大,澳大利亚和巴西。中国和俄罗斯的管理委员会资格正在确认过程中。世界卫生组织是该组织的观察员。IMDRF 管理委员会由成员国的监管官员组成,将会提供论坛来制定关于策略、政策、方向、会员资格及活动方面的指南。管理委员会每两年开一次会议,会议议程包括了一次面向所有相关方,包括工业界、学术界、医疗健康专业人士及消费者和病人组织的公开会议。IMDRF 的主席和秘书处采取年度轮换的方式。现在该组织处于良好的运作过程中。
1.7 亚洲协调工作组织 Asia Harmonization Work Party (AHWP)亚洲医疗器械法规协调组织(Asian Harmonization Working Party,AHWP)是由亚洲地区包括中国、韩国、菲律宾、新加坡、马来西亚、印度尼西亚、泰国、印度等 23 个经济体所组成之医疗器材法规调和组织,该组织于 1999 年成立,是亚洲各国医疗器械法规主管机构与业者推动法规协调的重要组织。AHWP 会议每年举行 1 次,由各国政府部门和行业协会轮流主办。其目的在于形成亚洲地区的医疗器械的法规协调的共同的方向,提供一个探讨和培训的平台,促进信息的交流。AHWP 积极地参与和开展各种活动,与法规主管部门和企业进行区域性或国际化的合作。例如,对 AHWP 成员现有的医疗器械法规的对比性研究;在 AHWP 协调医疗器械的定义、分类和术语;规范上市后警戒系统;通过培训提高水平;在东盟推进通用的申报资料等。
本文来源于:鸿远医疗器械咨询 http://www.yixiezixun.com
 在线客服
在线客服