

����ҽ����е��Ʒע�����ݡ�ҽ����е����Ŀ¼�����������Ĺ������Ϊ���࣬�������Ϊ17-07-07������ʽ��������������ϱ�����(Hawley������)��ѹĤ��������
����ע�����Ҫ��
����(һ)�����Ϣ
����1.�����
����1.1��Ʒ����
������Ʒ����Ӧ���Ʒ����һ�£���һ�����Ĵʺ�һ�㲻����������������ɡ�����������������������������������ʽ����������ժ��������
����1.2ע�ᵥԪ���ֵ�ԭ���ʵ��
����������ע�ᵥԪԭ�����Բ�Ʒ�ļ���ԭ�����ṹ��ɡ�����ָ������÷�ΧΪ�������ݡ�
�������磺��Ʒ�ṹ��ɡ�����ָ�겻ͬ��Hawley��������ѹĤ���������鲻��Ϊͬһע�ᵥԪ�걨��
����ĤƬ���ʲ�ͬ��ѹĤ���������鲻��Ϊͬһע�ᵥԪ�걨��
����2.�����д���б�(������)
����3.��Ʒ�б�
����4.�����ļ�
����5.ҽ����еע���걨ǰ���ܻ�������ϵ�����ͨ��¼(������)
����6.����������
����(��)��������
����1.����
����1.1��Ʒ��ͨ�����Ƽ���ȷ������
����1.2��Ʒ�Ĺ������
����1.3���÷�Χ
����1.4�����ã������й��걨��Ʒ�ı�����Ϣ�������ر�ϸ�ڣ��磺�걨��Ʒ����ʷ�����������ύ����Ϣ���������������в�Ʒ�Ĺ�ϵ�ȡ�
����2.��Ʒ����
����2.1��е������ԭ������
����2.1.1��Ʒ����ԭ��/���û���
���������ν�����ɺ����������˻ص���ʼλ�õ�����;���κ�������Χ�Ĺ������ڽ���֯��Ҫһ��ʱ����ɸĽ�(�������ƺ�6~12����������֯����ؽ�����������ά�ĸĽ���Ҫ1������[1])����������ƽ�⡢���Ŀ�ǻ����ϰ��;������������������ĥ���ȳ�������Ҳ��Ӱ������Ч���������Ҫ��ȡһ���Ĵ�ʩ���ڴ��ڼ佫����ά������������ۼ�����λ�ã���ֹ�����ƶ����ô�ʩ������ (retention)�������ǽ��ι��̲��ɻ�ȱ��һ����Ҫ�κ���ɲ���[2]��
�������û�е����װ��(������ retainer)���ɽ����ݻ����ȶ��ڽ��κ���ض�λ�ã������ٴ�����Ч������ֹ����(relapse)[3]��
����2.1.2��Ʒ�Ľṹ�����
������������Ҫ��ΪHawley��������ѹĤ����������ʽ��ͼ1��
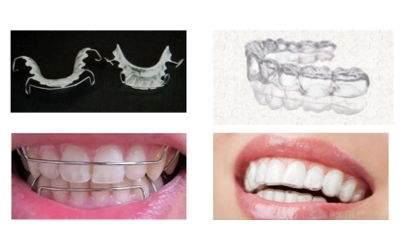
����ѹĤ������ͨ����ҽ�ü�������ϩ���۶Ա��������Ҷ�����(PET)���۶Ա��������Ҷ�����-1,4-��������״���(PETG)��۰����Ȳ��ϣ�����ѹ����ĥ������ɡ�
����2.2�ͺŹ��
����2.3��װ˵��
����2.4�����
����2.5��ͬ���/��ǰ����Ʒ�IJο��ͱȽ�
����3.���÷�Χ�ͽ���֤
����3.1���÷�Χ�����ڹ��������ν�����ɺ����Ч��
����3.2Ԥ��ʹ�û�������Ʒ�״�ʹ��ʱ�������ʵ�ҽ�ƻ�����רҵ��ҽΪ����װ�������Ժú���ɻ����ճ�����װ����������ҽ�����ڸ��
����3.3������Ⱥ������ƷĿ����ȺΪ��Ҫ��������������ɺ���Ч�Ļ��ߡ�
����3.4����֤��
����3.4.1����ȫ��ϵͳ�Լ�����������ǻר��ҽ����ȫ��ҽ���ۺ��������ʺ���������������;
����3.4.2�Ա��������Ϲ����ߡ�
����4.�걨��Ʒ������ʷ(������)
����(��)���ٴ�����
����1.��Ʒ���չ�������
����ҽ����еע����� ������Ӧ����YY/T 0316��ҽ����е���չ�����ҽ����е��Ӧ�á��Ա��������з��շ������Բ�Ʒ��������ȫ����ʵʩ���չ�������������YY/T 0316��¼E�о��˱���������ҪΣ������(����1)��
��1 ����������ҪΣ������
| Σ���ķ��� | Σ�����γ����� | ���ܵĺ�� | |
|---|---|---|---|
| ����ѧΣ�� | ���ﲻ������ |
���������ӹ��������IJ��ϣ����������õ����������ԡ� �Ƽ��������������²��Ϸ����仯���������õ����������ԡ� |
�������ԡ��̼��ȷ�Ӧ���������̼������ȡ� |
| ���Ϲ��� | ʹ��ǰδ�˽�߹���ʷ��δ��˵�����о�ʾ������ϵĽ����к�Ԫ�س��ꡣ | �����߹��� | |
| �ٸ�Ⱦ��/���Ⱦ | ʹ��ǰδ��Ч��ࡢ��������Ƶ��ϱ����ģ�Ͳ�һһ��Ӧ����ʶ���塣 | �����߾ֲ��������Ⱦ�� | |
|
��е Σ�� |
��Ʒ���治�⻬ | ��Ʒ�ֲڲ���ĥ�𡢻�����������ǻճĤ��������֯�ȡ� | �Ի�����������ǻ�Ĥ������֯��������˺��� |
| ���ʵ��IJ��ϻ�ӹ������й��ղ����Ŀ���ʧЧ������ѧ����������Ҫ�� | δѡ����ȷ��ԭ���ϻ��������̿���ʧЧ�� |
���������ѣ����ܵ��»������̡����ƻ��߿�ǻ������֯���Ի�������˺��� |
|
|
���� �� |
���ʵ��IJ���˵�� |
��1������δ��˵�����Ƽ��ķ�����ϴ�����±�����������ʴ���Ρ� ��2������δ��ʱ���δ��ʱ��������������������Ч�����ﵽԤ�ڡ� ��3�����߱����������о����ʻ��ɶ���δ��ʱ��ҽ�������Լ��ֶ������� ��4������δ��ҽ����˵����Ҫ�����õĿ�ǻ����ϰ�ߡ� |
��1������֤ʹ�õ���Ч�Ի�������ʹ�á� ��2�����������ѣ����ܵ��»������̡����ƻ��߿�ǻ������֯���Ի�������˺��� ��3������ȣ�������ܼ����ȿ�ǻ���⡣ |
|
���� �� |
����ƫ��Ԥ���Ļ������� | �������������¶ȡ�ʪ�ȡ����ȣ�������Ҫ�� | ��Ʒ��ɫ����ʴ�� |
| ����Ļ�е�ƻ� | ���ˡ�ʹ�ù����з�������Ļ�е���ƻ��� | ��Ʒʹ���������õ���֤��������֤����ʹ�á� | |
ҽ����е��Ʒע�����������ҵ��Ӧ����������Ʒ�ص�ȷ������Σ������ͨ����Ʒ��ƿ��ơ���Ʒԭ����ѡ��Ʒ��������ָ����ƶ�����ȷ�ı�ǩ��ʶ�������ͼ�����ơ���Ʒ˵����ȶ����ʩ�Խ��ͷ������ɽ���ˮƽ��
����2.ҽ����е��ȫ�����ܻ���ԭ���嵥
����3.��Ʒ����Ҫ���鱨��
����3.1�걨��Ʒ���ñ����
����3.2��Ʒ����Ҫ��
����ҽ����еע�������Ʒ����Ҫ����ƶ�Ӧ���ϡ�ҽ����е��Ʒ����Ҫ���дָ��ԭ�ķ���Ҫ��������Ӧ���ݲ�Ʒ�ļ����������ٴ�ʹ�������ȷ����Ʒ��ȫ��Ч�������ɿصļ���Ҫ��ͼ��鷽������ָ��ԭ�������������Ҫ���ǵIJ�Ʒ������������ָ�꣬�����˸���������Ʒ�ļ����ص����;�ƶ���Ӧ������ָ�꣬������ָ�겻�õ���ǿ���Թ��ұ�����ҵ����
����3.2.1Hawley��������Ʒ����ָ������Ӧ�������¼��㣺
����3.2.1.1��ƣ�Ӧ��ҽ�ƻ����ṩ�Ĺ���ģ�ͼ�����ļ����졣
����3.2.1.2ԭ���ϣ���������������Ӧʹ�þ���ҽ����еע��֤����ƾ֤��ԭ���ϣ���ʹ��δ��ע���ԭ���ϣ�Ӧ�г����õ�����ָ�꣬���������оۺ���Ӧ��YY/T 0270.2������ѧ ���оۺ��� ��2���֣��������оۺ���г����õ�����ָ�ꡣ
����3.2.1.3��ۣ���������¶�ڿ�ǻ�Ľ�������Ӧ�߶��⣬�����ֲڶ�Ӧ�ﵽRa≤0.025μm����λ�塢���С�������������ı���Ӧ�⻬���й��������ơ���϶��
����3.2.1.4��֯�棺����������֯�治�ô��ڲ���ʯ�ࡣ
����3.2.1.5ɫ�ȶ��ԣ�������֬����Ӧ��ɫ���ȣ��������õ�ɫ�ȶ��ԡ�
����3.2.1.6�ܺ϶ȣ�������Ӧ�빤��ģ�������ι̣��������ѡ�
����3.2.1.7����˿Ӧһ����ͣ���Ӧ�����Ե����ƺۼ���
����3.2.1.8����������֬���в������Ӧ��С��0.6mm��
����3.2.2ѹĤ��������Ʒ����ָ������Ӧ�������¼��㣺
����3.2.2.1��ƣ�Ӧ��ҽ�ƻ����ṩ�Ĺ���ģ�ͼ�����ļ����졣
����3.2.2.2ԭ���ϣ�ӦΪ����ҽ����еע��֤������Ƭ/����ĤƬ�Ƴɣ���ʹ��δ��ע���ԭ���ϣ�Ӧ�г����õ�����ָ�ꡣ
����3.2.2.3��Ʒ����Ҫ��
������ۣ���Ʒ�ڡ������ͱ�ԵӦ�⻬�����þ����������ʡ��۵㡢����/�졢���ݡ����⡢ë�̡���϶�����۵�ȱ�ݡ�
������ɫ��������Ӧ��ɫ�������и��Ի������������ҵ�����ơ�
��������: ѹĤ������Ӧ����ȫ�����У�����е���ĥ����Ӧ���Ǹ������в��֡�
�����ߴ�ȷ�ԣ��������빤��ģ�͵�ƽ���ߴ�ƫ��Ӧ≤0.5 mm��
����3.2.2.4�������ܣ�
�����ܶȣ�������2.6g/cm3��
������ȣ���Ʒ�ĺ��Ӧ�����Ƶķ�Χ��(��ȷ��Сֵ)��
������ˮֵ����������λ�������������(��ˮ��)��Ӧ����32µg/mm³��
�����������ܣ������������쵯��ģ��Ӧ����500MPa~2400MPa��Χ�ڡ�
������ĥ�����ܣ���Ʒ����¼�ķ���һ���飬������ʧӦС��0.25g/1000r��
������˺�����ܣ� ֱ��˺��ǿ��Ӧ����100kN/m��
�����ܽ�ֵ����������λ�����������ʧ(ˮ�ܽ���)��Ӧ����1.6µg/mm³��
�����ܺ϶ȣ�������Ӧ�빤��ģ�������ι̣���ģ���ϲ�Ӧ�����𡢰ڶ�����ת���³��Ȳ��ȶ�����
�������ȶ��ԣ���Ʒ����¼�ķ��������飬�����仯������1%��
����ɫ�ȶ���(������)��������Ӧ��ɫ���ȣ��������õ�ɫ�ȶ��ԡ�
����3.2.2.5��ѧ���ܣ�
�������ȣ�����Һ�Ϳհ�ҺpHֵ֮��Ӧ������1.5��
�����ؽ����ܺ���������Һ���ؽ����ܺ���Ӧ������1μg/ml��
������ԭ����(��������)������Һ�Ϳհ�Һ���ĸ��������Һ[ c (KMnO4)=0.002mol/L]�����֮��Ӧ������2.0ml��
����������������������������Ӧ������2mg��
������������ָ����������Ƭ/����ĤƬ���Ѿ���⣬����������֤�������ӹ�����Ը���ָ����Ӱ�죬�������ڳ�Ʒ���ظ���⡣
����3.3���鱨��
����3.3.1���鱨������������˰��ա�ҽ����еע���Լ�����涨�����ߵ��Լ챨�棬Ҳ������ί�������ʵ�ҽ����е����������ߵļ��鱨�档
����3.3.2ͬһע�ᵥԪ��������IJ�ƷӦ���ܹ�������ע�ᵥԪ��������Ʒ�İ�ȫ�Ժ���Ч�ԡ�
����4.�����
����ҽ����е��Ʒע��֤�����������걨�IJ�Ʒ���ṩ��Ӧ���о����ϡ�
����4.1��ѧ�����������о�
����ԭ������ȡ��ҽ����еע��֤��ƾ֤�����������ṩԭ���Ϲ�Ӧ����Ϣ�����ձ���
����ԭ�����������������з������ģ�����ȷ����ԭ�ϵĻ�ѧ���ơ�CAS�š������ƺš���ѧ�ṹʽ/����ʽ��������(������)����Դ�ʹ���(������)��ʹ��������ɱ��������á����ϵı��������˵����ձ�����ص�֧�������ϣ��������б�����ʽ�ṩ����˵��ԭ���ϵ�ѡ�����ݼ���Դ��
�����걨������Ӧ������Ʒ�����о������Լ���Ʒ����Ҫ����о��ͱ���˵������������ָ���ȷ�����ݣ������õı��������õ�ԭ�����ۻ�����
����Hawley����������ѧ���������Ƹ�˿��ҽ����װ������أ��ɲ����������о���
����ѹĤ����������ѧ�о���ά�ֽ���Ч�������ʶȵȷ���������ҪӰ�죬�������һЩ��������ѧ�о���Ŀ���о��������ο���
�����о���Ŀ�����ϵĶ�����ѧ���֣��絯��ģ��������ǿ�ȡ�����ǿ�ȡ�����Ӧ������伫�Ȳ���;���ϵij�����ѧ���֣���Ҫ��Ӧ���ɳ����ܼ��Բ�ͬ������ʴ�ĵֿ����ܡ�
�����о��������Բ�����ѧ���ܵ��о�ͨ������ר�����������������鼰�ɳ����飬�Ա�������ѧ���ֵ��о���Ҫ��ͨ������Ӧ����������ʵ��Ӧ��������������Ӧ��������ͨ��ʹ�õ�������Ԫ��ģ�⣬ʵ��Ӧ�������������ǵ�ⷨ��ģ�����鷨��
����4.2����ѧ�����о�
�����������Ӵ���ǻ�Ĥ���������Ӳ��֯����棬���ڱ���Ӵ�����е������YY/T 0268������ѧ��ǻҽ����е����ѧ���۵�1��Ԫ�����������顷���Ӵ�ʱ���ϲ�Ʒ�����ʹ�÷���ȷ����Ӧ�����ۼ�ʹ��ʱ�䣬��������������Ӧ��ѭGB/T 16886.1��ҽ����е����ѧ���۵�1���֣����չ��������е����������顷��YY/T 0268������ѧ��ǻҽ����е����ѧ���۵�1��Ԫ�����������顷���Ҫ��
��������ʹ����ȡ��ҽ����еע��֤ԭ����������Hawley�������ɻ�������ѧ���飬����ע��֤��Ϊ���������������о����ϵ�һ���֡�
��������ʹ����ȡ��ҽ����еע��֤ԭ����������ѹĤ����������������ѧ�����������Ӧ�ṩ��Ʒ�������ӹ����ԭ��������������ȫ��Ӱ����о����ϡ�
����������˵����������ѧ��������ɻ�ʹ��δȡ��ҽ����е��Ʒע��֤��ԭ���ϣ�Ӧ����DZ�ڵ��ۼ����ã��ɸ���YY/T 0268������ѧ��ǻҽ����е����ѧ���۵�1��Ԫ��������������Ŀѡ��GB/T 16886.1��ҽ����е����ѧ���۵�1���֣����չ��������е����������顷�����Ӵ�����ʱ�����۳�Ʒ������ѧ������Ŀ�������ٿ��ǣ�ϸ�����ԡ��ٷ��ͳ�����Ӧ���̼���Ƥ�ڷ�Ӧ��������ȫ�����ԡ��Ŵ����ԡ�
����4.3��ࡢ����������о�
����������ͨ����ʹ������ࡢ����(������)��Ӧ����ȷ�Ƽ�����ϴ����������(������)�����յ�ȷ�������Լ���֤������о����ϡ�������������������ܲ�����������,Ӧ����������IJ�Ʒ���в������Ե��о�����ȷ��������Ϣ����ȡ�Ĵ������������ṩ����о����ϡ�
����4.4֤����Ʒ��ȫ�ԡ���Ч�Ե������о����ϡ�
����5.���ٴ�����
����6.�ȶ����о�
����6.1Ӧ��ȷ��������Ʒ�İ�װ��Ч�ڣ����ڻ��߿�ǻ��ȡģ����װ������ޡ���Ӧ��ȷ��Ʒ���ʱ��ڡ�
����6.2�����ȶ��ԣ�Ӧ��ȷ��Ʒ���ڰ�װ��ʽ��ȷ����װ�����Ƶ����䴢�������£��ڲ�Ʒ��Ч�����ܹ��Բ�Ʒ�������ò����ֲ�Ʒ��ࡣ��Ʒ��װ��֤�ɲο�GB/T 19633.1��YY/T 0681.15�ȱ����У��ύ��Ʒ�İ�װ��֤���档��װ���ϵ�ѡ��Ӧ�����������أ���װ���ϵ�������ѧ����;�Ƿ�������ؽ������������������;��װ��������ͺ��ܷ���̵���Ӧ��;��װ�������Ʒ����Ӧ��;��װ�������ǩϵͳ����Ӧ��;��װ����������������̵��ʺ���;��װ��Ч�ڡ���װ��֤����������Ӧ���װ˵���и�������Ϣ�����
����(��)�ٴ���������
����ҽ����еע��֤����ע��������Ӧ���ա�ҽ����е�ٴ����ۼ���ָ��ԭ��ѡ���ٴ�����·�����ύ�ٴ��������ϡ�
����1.ͨ��ͬƷ��ҽ����е�ٴ����ݽ��з���������
�������ա�ҽ����е�ٴ����۵�ͬ����֤����ָ��ԭ����Ʒ��������ͬƷ����е���жԱȣ���ѹĤ���������������ܡ���˺�����ܵ������ѧ����ָ�꣬�����˿�ͨ��ʵ���öԱ����ݣ����ͨ��ԭ���ϼ��鱨����ʵ�����ݡ�ֵ��ע����ǣ��걨��Ʒ�ͶԱȲ�Ʒ������������һ�¡�
�������ڲ�������ָ�꣬����/��ҵ����ISO�����ƶ��˽�����ֵ�����ձ��еķ����Ͳ������в��ԣ�������ϱ�������ֵҪ��һ�㲻��Ҫ����ͬƷ����е���в��ԶԱȡ����磺YY/T 0270.2������ѧ ���оۺ��� ��2���֣��������оۺ����YY/T 0114��ҽ����Һ����Ѫ��ע�������þ���ϩר���ϡ�(������)����ر�������ȷ������ֵ������Ҫ��ɲ����жԱȣ��걨��Ʒ���鱨���е�ʵ��ֵ������ر�Ҫ�ɣ�����ˮֵ���ܽ�ֵ�����ȡ��ؽ����ܺ�������ԭ���ʡ�����������
������ѡ���ͬƷ�ֲ�Ʒ���걨��Ʒ�ڲ�����Ŀ�����в��죬���ۺ���������Ӧ������ͬ���Ա���Ŀ��ͬƷ����Ϣ����ȡ�ģ����������ա�ԭ�����ƺŵȣ�Ҳ�ɲ���дֱ����Ϊ������ṩ��ƥ��İ�ȫ��Ч��֤�ݡ�
����2.ͨ���ٴ��������ݽ��з���������
���������ٴ��������ϡ��ٴ����ݲ�����ȷ�ϱ�������ȫ����Ч�ģ�Ӧ��չ�ٴ����顣���й����ڿ�չ���ٴ�����ģ�����ϡ�ҽ����е�ٴ��������������淶������ӦҪ��;�ھ��չ�ٴ�����ģ�����ϡ�����ҽ����е�����ٴ��������ݼ���ָ��ԭ����ӦҪ��
����(��)ҽ����еע��֤������Ʒ˵����ͱ�ǩ����
������Ʒ˵����ͱ�ǩҪ��ı�дӦ���ϡ�ҽ����е˵����ͱ�ǩ�����涨������ر���Ҫ��ͬʱ��Ӧע�����¼���(�����ڴ�)��
����1.ԭ���������Ϣ��
����2.����ÿ�����ʱ�䡣
����3.��Ʒʹ��˵��������֤������Ӧ��˵��������ȡ�
����4.ע������Ӧ��ȷ�������ݣ�
����4.1��Ҫ�ھ���רҵ���ʵ�ҽʦָ���½����״������
����4.2����������ǰӦ����������������ݲ�Ʒ�IJ���������ȷ�Ƽ����������������
����4.3������������Ҫ�����ʾ�����ԡ�
����4.4��ʳ��ˢ����Ҫ��
����4.5������Ӧ�Ĵ���������
����4.6�����桢��������е�Ҫ��
����5.����֤����Ӧ���������ݣ�
����5.1����ȫ��ϵͳ�Լ�����������ǻר��ҽ����ȫ��ҽ���ۺ��������ʺ���������������;
����5.2�Ա��������Ϲ����ߡ�
����(��)ҽ����еע��֤�걨����������ϵ�ļ�
����1.����
����2.����������Ϣ
����3.����������ϵ����
����4.����ְ�����
����5.��Դ��������
����6.��Ʒʵ�ֳ���
����7.����������ϵ�IJ����������Ľ�����
����8.����������ϵ������Ϣ
����9.����������ϵ�˲��ļ�
��Զҽ����е��ѯ����˾ ��һ�Ҽ���רҵ��ҽ����е��ѯ����˾��רע�ṩȫ�������磺���ڡ����ݡ���ݸ����ɽ����ɽ�����ݡ�˳�¡��������Ϻ������������졢�ɶ������ա����ա��㽭��֪�����е�ҽ����е��������ѯ����Զҽ����е��ѯרҵ�����ڣ�ҽ����е��Ʒע��֤��������ѯ��ҽ����е��Ʒ����綨������,����ҽ����е��������֤��һ��ҽ����е��Ʒ�������졢��������Լ�ע�ᡢ����ҽ����еע�ᡢҽ����е��Ӫ����֤���졢����ҽ����е��Ӫ������CE��֤��ISO13485��֤��FDAע�ᡢFDA��֤���ٴ����顢ҽ����е����������ϵ��֤����ϵ���������ȷ���ļ�����( ISO13485, GMP, CE��QSR820��CMDCAS);ע�ᡢҽ����е��������֤������Ʒ����Ҫ���ƶ��������ļ���ͬ���Ʒ�Ա����ٴ��������ϱ�д��ע�����ϱ�д������ҽ����е��������걨����ż������ĵ��ṩһվʽ����˾����ӭ����ѯ�������
 ���߿ͷ�
���߿ͷ�