

医疗器械注册,是指依照法定程序,对拟上市销售、使用的医疗器械的安全性、有效性进行系统评价,以决定是否同意其销售、使用的过程。医疗器械是指直接或者间接用于人体的仪器、设备、器具、体外诊断试剂及校准物、材料以及其他类似或者相关的物品,包括所需要的计算机软件。效用主要通过物理等方式获得,不是通过药理学、免疫学或者代谢的方式获得,或者虽然有这些方式参与但是只起辅助作用。目的是疾病的诊断、预防、监护、治疗或者缓解;损伤的诊断、监护、治疗、缓解或者功能补偿;生理结构或者生理过程的检验、替代、调节或者支持;生命的支持或者维持;妊娠控制;通过对来自人体的样本进行检查,为医疗或者诊断目的提供信息。为了规范管理国家对医疗器械按照风险程度实行分类管理。
第一类是风险程度低,实行常规管理可以保证其安全、有效的医疗器械。
第二类是具有中度风险,需要严格控制管理以保证其安全、有效的医疗器械。
第三类是具有较高风险,需要采取特别措施严格控制管理以保证其安全、有效的医疗器械。
第一类医疗器械实行产品备案管理,第二类、第三类医疗器械实行产品注册管理。医疗器械注册人、备案人应当加强医疗器械全生命周期质量管理,对研制、生产、经营、使用全过程中医疗器械的安全性、有效性依法承担责任。
第一类医疗器械产品备案和申请第二类、第三类医疗器械产品注册,应当提交下列资料:
(一)产品风险分析资料;
(二)产品技术要求;
(三)产品检验报告;
(四)临床评价资料;
(五)产品说明书以及标签样稿;
(六)与产品研制、生产有关的质量管理体系文件;
(七)证明产品安全、有效所需的其他资料。
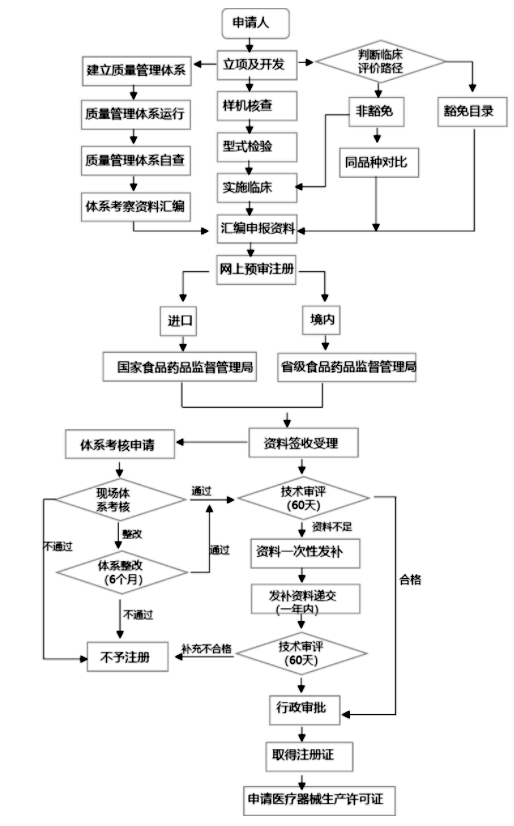
进口及国内医疗器械注册流程图
申请第二类医疗器械产品注册,注册申请人应当向所在地省、自治区、直辖市人民政府药品监督管理部门提交注册申请资料。
向我国境内出口第二类、第三类医疗器械的境外注册申请人,由其指定的我国境内企业法人向国务院药品监督管理部门提交注册申请资料和注册申请人所在国(地区)主管部门准许该医疗器械上市销售的证明文件。未在境外上市的创新医疗器械,可以不提交注册申请人所在国(地区)主管部门准许该医疗器械上市销售的证明文件。
国务院药品监督管理部门应当对医疗器械注册审查程序和要求作出规定,并加强对省、自治区、直辖市人民政府药品监督管理部门注册审查工作的监督指导。
产品检验报告应当符合国务院药品监督管理部门的要求,可以是医疗器械注册办理申请人、备案人的自检报告,也可以是委托有资质的医疗器械检验机构出具的检验报告。
鸿远医疗器械咨询服务公司 是一家技术专业的医疗器械咨询服务公司,专注提供全国各地如:深圳、广州、东莞、中山、佛山、潮州、顺德、广西、上海、西安、重庆、成都、安徽、江苏、浙江等知名城市的医疗器械领域技术咨询服务。鸿远医疗器械咨询专业服务于:医疗器械注册证代办理咨询、医疗器械产品分类界定代办理,代办医疗器械生产许可证、一类医疗器械产品备案代办、体外诊断试剂注册、进口医疗器械注册、医疗器械经营许可证代办、二类医疗器械经营备案、CE认证、ISO13485认证、FDA注册、FDA认证、临床试验、医疗器械质量管理体系认证及体系建立与过程确认文件建立( ISO13485, GMP, CE,QSR820,CMDCAS);注册、医疗器械出口销售证明、产品技术要求制订、技术文件、同类产品对比免临床评价资料编写、注册资料编写辅导、医疗器械广告批文申报、电磁兼容整改等提供一站式服务公司,欢迎您咨询与合作!
 在线客服
在线客服