

进口医疗器械注册咨询据CFDA发布的《国家药监局发布药品医疗器械境外检查管理规定》为进一步规范药品医疗器械境外检查工作,保证进口药品医疗器械质量,国家药监局2018年12月28日发布《药品医疗器械境外检查管理规定》(以下简称《规定》)。
境外检查工作是国际通行监管方式,也是我国药品医疗器械监管工作迈向国际化的重要一步,对督促境外企业符合中国法规起到重要作用,越来越受到国际社会的关注与肯定。
为落实2018年4月12日国务院常务会议中“关于加强进口药品境外生产现场检查”的要求,保障我国人民群众的用药用械安全,进一步规范境外检查工作,国家药监局总结近年来药品医疗器械境外检查工作经验,同时参考了其他国家药品监管机构的相关文件,起草了《规定》。《规定》的出台,既强化了我国对于境外药械的监管,也实现与国际通行监管方式接轨。
《规定》明确,境外检查是针对已在中华人民共和国境内上市或者拟在境内上市的药品和医疗器械;境外检查不限于生产现场检查,而延展为境外研发及生产场地检查。检查任务的形成,是考虑药品医疗器械的注册审评审批、监督检查、检验、投诉举报、不良反应监测等多渠道风险因素,体现风险防控管理要求。
检查结果综合评定采取风险评估的原则,综合考虑缺陷的性质、严重程度以及所评估产品的类别判定检查结果,评定为符合要求、整改后符合要求或不符合要求。《规定》将拖延、阻碍、限制或拒绝检查等情形直接判定为“不符合要求”。
检查结果处理区分了风险控制手段和立案调查处理两种情形。对于检查发现的严重质量风险的,将立即采取风险控制措施。对于采取风险控制措施的,风险排除后,被检查单位可以向国家药监局申请解除风险控制措施。在检查中发现涉嫌违法违规的,组织依法立案调查处理。
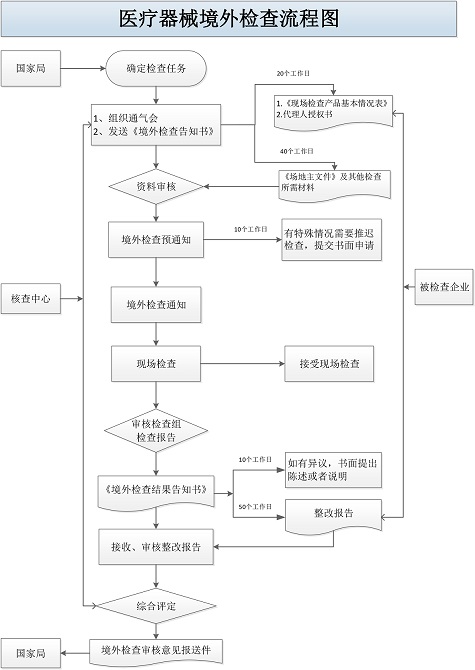
进口医疗器械注册境外生产公司现场检查流程图:
鸿远医疗器械咨询服务公司 是一家技术专业的医疗器械咨询服务公司,专注提供全国各地如:深圳、广州、东莞、中山、佛山、潮州、顺德、上海、西安、重庆、成都、安徽、江苏、浙江等知名城市的医疗器械领域技术咨询服务。鸿远医疗器械咨询专业服务于:医疗器械产品注册证代办理咨询、医疗器械产品分类界定代办理,代办医疗器械生产许可证、一类医疗器械产品备案代办、体外诊断试剂注册、进口医疗器械注册代办、医疗器械经营许可证代办、二类医疗器械经营备案、CE认证、ISO13485认证、FDA注册、FDA认证、临床试验、医疗器械质量管理体系认证及体系建立与过程确认文件建立( ISO13485, GMP, CE,QSR820,CMDCAS);注册、医疗器械出口销售证明、产品技术要求制订、技术文件、同类产品对比免临床评价资料编写、注册资料编写辅导、医疗器械广告批文申报、电磁兼容整改等提供一站式服务公司,欢迎您咨询与合作!
 在线客服
在线客服