

ЎЎЎЎТҪБЖЖчРөЧўІбөНЖөЦОБЖТЗІъЖ·өДГьГыУҰ·ыәПЎ¶ТҪБЖЖчРөНЁУГГыіЖГьГы№жФтЎ·(№ъјТКіЖ·Т©Ж·ја¶Ҫ№ЬАнЧЬҫЦБоөЪ19әЕ)Ј¬ІЙУГЎ¶ДҝВјЎ·»т№ъјТұкЧјЎўРРТөұкЧјЙПөДНЁУГГыіЖЎЈІъЖ·ГыіЖУҰТФМеПЦІъЖ·өД№ӨЧчФӯАнЎўјјКхҪб№№МШХчЎў№ҰДЬКфРФОӘ»щұҫЧјФтЎЈ
ЎЎЎЎИз“өНЖөЦОБЖТЗ”Ўў“ЙсҫӯәНјЎИвҙМјӨЖч”Ўў“өНЦЬІЁЦОБЖТЗ”Ўў “өзХлЦОБЖТЗ”Ўў“өзЧУХлБЖТЗ”өИЎЈ
ЎЎЎЎТҪБЖЖчРөөНЖөөзБЖТЗІъЖ·өДҪб№№әНЧйіЙУҰёщҫЭІъЖ·ЧФЙнМШөгИ·¶ЁҪб№№ЧйіЙЈ¬НЁіЈУЙЦч»ъ(РЕәЕІъЙъј°ҝШЦЖЧ°ЦГ)ЎўөзФҙЧ°ЦГ(ДЪЦГ»тНвЦГ)Ўўөзј«ј°ЖдЛыёҪКфІҝјюЧйіЙЎЈ
ЎЎЎЎТҪБЖЖчРөІъЖ·ЧўІбЙкЗлИЛУҰГиКцЙиұёЛщУРЧйјюј°ёҪјюЈ¬УҰМṩІъЖ·Ҫб№№ЧйіЙөДКөОпНјЖ¬»тКҫТвНјЎЈ¶ФУЪ¶аЦЦРНәЕ№жёсөДІъЖ·УҰБРұнЛөГчёчРНәЕЦ®јдөДЛщУРТмН¬ЎЈ
ЎЎЎЎИзЙиұёҫЯУРРиТӘЧйәПК№УГөДЖдЛыЙиұё(ИзУРөзЖш»тХЯНЁРЕБ¬ҪУөДЙиұё)Ј¬УҰМṩҪУҝЪЙијЖЛөГчЈ¬ТФј°¶ФУҰөДЧйәПК№УГЙиұёөДПкПёЛөГчЎЈ
ЎЎЎЎІъЖ·ЧйіЙКҫАэЈә
ЎЎЎЎНј1 ІъЖ·КҫАэНј
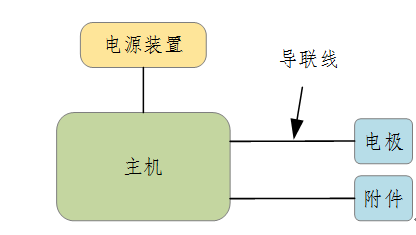
ІъЖ·өДЦчТӘ·зПХ
ЎЎЎЎТҪБЖЖчРөЧўІб°мАнөНЖөөзБЖТЗ·зПХЧКБПұаРҙЦчТӘІОҝјYY/T 0316-2016Ў¶ТҪБЖЖчРө·зПХ№ЬАн¶ФТҪБЖЖчРөөДУҰУГЎ·ЎЈ·зПХ№ЬАн»о¶ҜТӘ№бҙ©ІъЖ·ЙијЖЎўЙъІъЎўЙПКРәуК№УГј°ІъЖ·өДХыёцЙъГьЦЬЖЪЎЈТӘМеПЦЧўІбЙкЗлИЛ·зПХ№ЬАн»о¶ҜјЖ»®өДНкХыРФЈ¬УИЖдЙПКР№ЬАнөД·зПХ·ЦОцУлЖАјЫ№эіМЎЈ¶ФУЪЙПКРЗ°·зПХ№ЬАнЦРЙРОҙИПЦӘөД·зПХЈ¬УҰФЪЙПКРәуҝӘХ№РЕПўКХјҜЈ¬Т»ө©·ўПЦТміЈј°КұҪшРР·зПХЖАјЫЈ¬ІЙИЎҝШЦЖҙлК©Ј¬ёьРВ·зПХ№ЬАнОДјюЎЈ
ЎЎЎЎөНЖөөзБЖТЗ·зПХ·ЦОцУҰІОҝјYY/T 0316-2016Ў¶ТҪБЖЖчРө·зПХ№ЬАн¶ФТҪБЖЖчРөөДУҰУГЎ·РРТөұкЧјПа№ШТӘЗуЈ¬ЦрТ»ҪшРР»ШҙрЈ¬ТІҝЙТФУГБРұнөД·ҪКҪБРКҫЎЈКЈУа·зПХ·ЦОцКұЈ¬Т»¶ЁТӘ·ЦОцЦрТ»ІЙИЎ·зПХҝШЦЖҙлК©ә󣬻᲻»бТэИл»тФміЙёьҙуөД·зПХЈ¬ИфТэИлРВөД·зПХЈ¬Ц»УРРВТэИл·зПХДЬЧӘ»ҜОӘҝЙҪУКЬ·зПХЈ¬·ҪДЬИПОӘ·зПХКЬҝШЎЈөНЖөөзБЖТЗұШРлҪшРР·зПХУлКХТж·ЦОцЈ¬КХТжҙуУЪ·зПХКұ·ҪҝЙҪУКЬЎЈ
ЎЎЎЎТҪБЖЖчРөЧўІбЦӨЙкЗлИЛМṩөНЖөөзБЖТЗЙПКРЗ°·зПХ№ЬАнұЁёжЈ¬ҙЛұЁёжЦјФЪЛөГчІўіРЕөЈә
ЎЎЎЎ——·зПХ№ЬАнјЖ»®ТСұ»ХэИ·өШКөК©ЎЈ
ЎЎЎЎЧЫәПКЈУа·зПХКЗҝЙҪУКЬөДЎЈ
ЎЎЎЎ——ТСУРЗЎөұ·Ҫ·Ё»сөГУлЧўІбЙкЗлИЛЙкұЁөДөНЖөөзБЖТЗІъЖ·Па№ШәНіці§әуБчНЁУлБЩҙІУҰУГөДРЕПўЎЈ
ЎЎЎЎУҰЛж·зПХ№ЬАнұЁёжТ»ІўёҪЙП°ьАЁ·зПХ·ЦОцЎў·зПХЖАјЫЎў·зПХҝШЦЖёЕКц№ЬАнЧКБПЎЈЦБЙЩУҰ°ьАЁЈә
ЎЎЎЎ——ІъЖ·°ІИ«МШХчЗеөҘ;
ЎЎЎЎ——ІъЖ·ҝЙФӨјыОЈПХ(Фҙ)ј°·ЦОцЗеөҘ(ЛөГчОЈПХ(Фҙ)ЎўҝЙФӨјыКВјюРтБРЎўОЈПХЗйҝцәНҝЙДЬ·ўЙъөДЙЛәҰЦ®јдөД№ШПө);
ЎЎЎЎ——·зПХЖАјЫЎў·зПХҝШЦЖҙлК©ТФј°КЈУа·зПХЖАјЫ»гұЁұнЎЈ
ЎЎЎЎ¶ФУЪ·зПХ·ЦОцәН№ЬАнёЕКцЈ¬УҰ°ьАЁТ»·Э·зПХЧЬҪбЈ¬ТФј°ИзәОҪ«·зПХҝШЦЖФЪҝЙҪУКЬіМ¶ИөДДЪИЭЎЈҙУЙъОпС§ОЈПХ(Фҙ)Ўў»ъРөОЈПХ(Фҙ)ЎўДЬБҝОЈПХ(Фҙ)ЎўУР№ШК№УГөДОЈПХ(Фҙ)ЎўРЕПўОЈПХ(Фҙ)әНО¬»ӨІ»ЦЬј°АП»ҜТэЖрөДОЈПХ(Фҙ)өИ·ҪГжЈ¬¶ФІъЖ·ҪшРРИ«Гж·ЦОцІўІыКцПаУҰөД·А·¶ҙлК©ЎЈ
ЎЎЎЎ1.·зПХ·ЦОц·Ҫ·Ё
ЎЎЎЎ1.1ФЪ¶Ф·зПХөДЕР¶Ёј°·ЦОцЦРЈ¬ТӘҝјВЗәПАнөДҝЙФӨјыөДЗйҝцЈ¬°ьАЁЈәХэіЈК№УГМхјюПВәН·ЗХэіЈК№УГМхјюПВЎЈ
ЎЎЎЎ1.2·зПХЕР¶Ёј°·ЦОцУҰ°ьАЁЈә¶ФУЪ»јХЯөДОЈПХ(Фҙ)Ўў¶ФУЪІЩЧчХЯөДОЈПХ(Фҙ)әН¶ФУЪ»·ҫіөДОЈПХ(Фҙ)ЎЈ
ЎЎЎЎ1.3·зПХРОіЙөДіхКјФӯТтУҰ°ьАЁЈәИЛОӘТтЛШЈ¬ІъЖ·Ҫб№№өДОЈПХ(Фҙ)Ј¬ФӯІДБПОЈПХ(Фҙ)Ј¬ЧЫәПОЈПХ(Фҙ)Ј¬»·ҫіМхјюЎЈ
ЎЎЎЎ1.4·зПХЕР¶Ёј°·ЦОцҝјВЗөДОКМв°ьАЁЈәЙъОпПаИЭРФОЈПХ(Фҙ);»ъРөОЈПХ(Фҙ);ДЬБҝОЈПХ(Фҙ);ІЩЧчРЕПўЈ¬°ьАЁҫҜКҫРФУпСФЎўЧўТвКВПоТФј°К№УГ·Ҫ·ЁөДЧјИ·РФ;К№УГ№эіМҝЙДЬҙжФЪөДОЈПХ(Фҙ)өИЎЈ
ЎЎЎЎ2.·зПХ·ЦОцЗеөҘ
ЎЎЎЎөНЖөөзБЖТЗөД·зПХ№ЬАнұЁёжУҰ·ыәПYY/T 0316-2016Ў¶ТҪБЖЖчРө·зПХ№ЬАн¶ФТҪБЖЖчРөөДУҰУГЎ·өДУР№ШТӘЗуЈ¬ЙуІйТӘөг°ьАЁЈә
ЎЎЎЎ2.1ІъЖ·¶ЁРФ¶ЁБҝ·ЦОцКЗ·сЧјИ·(ТАҫЭYY/T 0316-2016Ў¶ТҪБЖЖчРө·зПХ№ЬАн¶ФТҪБЖЖчРөөДУҰУГЎ·ёҪВјC)ЎЈ
ЎЎЎЎ2.2ОЈПХ(Фҙ)·ЦОцКЗ·сИ«Гж(ТАҫЭYY/T 0316-2016Ў¶ТҪБЖЖчРө·зПХ№ЬАн¶ФТҪБЖЖчРөөДУҰУГЎ·ёҪВјE)ЎЈ
ЎЎЎЎ2.3·зПХҝЙҪУКХЧјФтЈ¬ҪөөН·зПХөДҙлК©ј°ІЙИЎҙлК©әу·зПХөДҝЙҪУКХіМ¶ИЈ¬КЗ·сУРРВөД·зПХІъЙъЎЈ
ЎЎЎЎТҪБЖЖчРөЧўІбҙъ°мёщҫЭYY/T 0316-2016Ў¶ТҪБЖЖчРө·зПХ№ЬАн¶ФТҪБЖЖчРөөДУҰУГЎ·ёҪВјE¶ФёГІъЖ·ТСЦӘ»тҝЙФӨјыөД·зПХҪшРРЕР¶ЁЈ¬өНЖөөзБЖТЗІъЖ·ФЪҪшРР·зПХ·ЦОцКұЦБЙЩУҰ°ьАЁТФПВөДЦчТӘОЈПХ(Фҙ)Ј¬ЧўІбЙкЗлИЛ»№УҰёщҫЭЧФЙнІъЖ·МШөгИ·¶ЁЖдЛыОЈПХ(Фҙ)ЎЈХл¶ФІъЖ·өДёчПо·зПХЈ¬ЧўІбЙкЗлИЛУҰІЙИЎУҰ¶ФҙлК©Ј¬И·ұЈ·зПХҪөөҪҝЙҪУКЬөДіМ¶ИЎЈ
ЎЎЎЎ3.ІъЖ·өДЦчТӘОЈПХ(Фҙ)
ЎЎЎЎ3.1ДЬБҝОЈПХ(Фҙ)
ЎЎЎЎөз»чОЈПХ(Фҙ)ЈәұЈ»ӨҪУөШЧиҝ№Ј¬ҪУөШІ»БјЈ¬¶ФөШЧиҝ№ҙу;»јХЯВ©өзБчЎўНвҝЗВ©өзБчі¬ұк;ПөНіөзҪйЦКҫшФөЗҝ¶ИІ»№»;УҰУГІҝ·ЦУлҙшөзІҝ·ЦГ»УРід·ЦёфАл;ЙиұёөДөзФҙІеН·КЈУаөзС№№эёЯ;»ъЖчНвҝЗөД·А»ӨХЦ·вұХІ»Бј;ЙиұёГ»УРЧг№»өДНвҝЗ»ъРөЗҝ¶ИәНёХ¶И;Тт№эҙуөДөзБчГЬ¶ИЈ¬ФміЙөДөзҙМјӨЙЛәҰЎЈ
ЎЎЎЎЙПКцЗйҝцөДіцПЦҝЙФміЙ¶ФК№УГХЯ»т»јХЯөДөз»чОЈПХ(Фҙ)ЎЈ
ЎЎЎЎөзіШОЈПХ(Фҙ)ЈәөзВ··ўЙъ¶МВ·Ј¬ҝЙДЬТэ·ў·ўИИЎўұ¬ХЁөИОЈПХ(Фҙ)ЎЈ
ЎЎЎЎөзҙЕДЬЈәҝЙДЬ№ІН¬К№УГөДЙиұё(јЖЛг»ъЎўҙтУЎ»ъЎўТЖ¶Ҝөз»°ЎўөзҙЕВҜЎўОўІЁВҜөИ)¶ФөНЖөөзБЖТЗөДөзҙЕёЙИЕЈ¬ҫІөз·Еөз¶ФөНЖөөзБЖТЗІъЙъөДёЙИЕЈ¬өНЖөөзБЖТЗІъЙъөДөзҙЕіЎ¶ФҝЙДЬ№ІН¬К№УГөДЙиұёөДУ°ПмөИТэ·ўөДОЈПХ(Фҙ)ЎЈ
ЎЎЎЎ3.2ЙъОпС§әН»ҜС§ОЈПХ(Фҙ)
ЎЎЎЎЙъОпС§Јә№«№ІіЎЛщОҙҫӯЗеПҙЎўПы¶ҫөДУлИЛМеҪУҙҘөДІҝјюТэЖрөДҪ»ІжёРИҫ;өНЖөөзБЖТЗөДФӯІДБПУР¶ҫУРәҰ¶ФИЛМеФміЙөДОЈПХ(Фҙ)ЎЈ
ЎЎЎЎ»ҜС§ЈәК№УГөДЗеҪајБЎўПы¶ҫјБІРБфТэ·ўөДОЈПХ(Фҙ)ЎЈ
ЎЎЎЎ3.3ІЩЧчОЈПХ(Фҙ)
ЎЎЎЎі¬іц°мАнТҪБЖЖчРөЧўІбЙкЗлИЛ№ж¶ЁөДКЩГьЖЪПЮК№УГЈ¬ҝЙДЬФміЙІЎИЛ»тК№УГХЯОЈПХЎЈ
ЎЎЎЎФЪЧўІбЙкЗлИЛ№ж¶ЁөДК№УГ»·ҫіМхјюНвК№УГІъЖ·Ј¬ҝЙДЬФміЙІъЖ·өДЛр»ө»тОЮ·ЁХэіЈ№ӨЧчЈ¬ІъЖ·КЩГьҪөөНЎЈ
ЎЎЎЎФЪЧўІбЙкЗлИЛ№ж¶ЁөДЦьҙж»·ҫіМхјюНвЦьҙжІъЖ·Ј¬ҝЙДЬФміЙІъЖ·өДЛр»ө»тОЮ·ЁХэіЈ№ӨЧчЈ¬ІъЖ·КЩГьҪөөНЎЈ
ЎЎЎЎЧ№ВдЈә№ӨЧчЧҙМ¬ЦРТЖ¶ҜІъЖ·»тІЩЧчІ»өұөјЦВІъЖ·Ч№ВдЎЈ(ККУГКұ)
ЎЎЎЎ3.4РЕПўОЈПХ(Фҙ)
ЎЎЎЎ°ьАЁұкјЗИұЙЩ»тІ»ХэИ·Ј¬ұкјЗөДО»ЦГІ»ХэИ·Ј¬І»ДЬұ»ХэИ·өШК¶ұрЈ¬І»ДЬУАҫГМщАОәНЗеіюТЧИПЎЈ
ЎЎЎЎТҪБЖЖчРөІъЖ·ЧўІбҙъ°мІ»·ыәП·Ё№жј°ұкЧјөДЛөГчКйЈ¬ұИИзЛөГчКйЦРОҙ¶ФПЮЦЖід·ЦёжЦӘЈ¬Оҙ¶ФІ»ХэИ·өДІЩЧчЎўУлЖдЛыЙиұё№ІН¬К№УГКұТЧІъЙъөДОЈПХ(Фҙ)ҪшРРҫҜёжЈ¬ОҙХэИ·ұкКҫҙўҙжМхјюЎўПы¶ҫ·Ҫ·ЁЎўО¬»ӨРЕПўЈ¬Оҙ¶ФТтіӨЖЪК№УГІъЙъ№ҰДЬЙҘК§¶шҝЙДЬТэ·ўөДОЈПХ(Фҙ)ҪшРРҫҜёжЈ¬Оҙ¶ФәПАнҝЙФӨјыөДОуУГҪшРРҫҜёжөИТэ·ўөДОЈПХ(Фҙ)ЎЈ
ЎЎЎЎ3.5ИнјюОЈПХ(Фҙ)
ЎЎЎЎІ»ХэИ·өДИнјюҝШЦЖЧҙМ¬ҝЙДЬФміЙКдіцВціеөзБч№эҙуЎЈіцПЦ¶ПөзЎў·ЗХэіЈ№Ш»ъөИЗйҝцЎЈ
ЎЎЎЎ№эУЪёҙФУөДҪзГжЙиЦГ»т·ЗФӨЖЪКдИлөјЦВІЩЧчТЧіцПЦҙнОуЎЈ
ЎЎЎЎИнјюұ»ЛжТвёД¶Ҝ»тТт°ІЧ°ЖдЛыИнјюЈ¬ҝЙДЬөјЦВЙиұёОЮ·ЁХэіЈ№ӨЧчЎЈ
ЎЎЎЎ3.6ТЖ¶ҜТҪБЖЖчРөәННшВз°ІИ«ОЈПХ(Фҙ)(ИзККУГ)
ЎЎЎЎТЖ¶ҜјЖЛгЦХ¶ЛөД·зПХЦчТӘұнПЦОӘПФКҫЖБіЯҙзРЎЎў·ЦұжВКөНЎўББ¶ИөНЈ¬КЬ»·ҫі№вУ°ПмҙуЈ¬өзіШИЭБҝРЎЈ¬КэҫЭҙ«КдК§ХжөИЈ¬І»ДЬ№»ВъЧгИ«ІҝБЩҙІТӘЗуЎЈ
ЎЎЎЎНЁУГЦХ¶ЛУлЧЁУГЦХ¶ЛПаұИЙијЖУГНҫІў·ЗУГУЪТҪБЖДҝөДЈ¬РФДЬЦёұкІ»ДЬ№»ВъЧгИ«ІҝБЩҙІТӘЗуЈ¬Н¬КұНЁУГЦХ¶ЛөДИнјюФЛРР»·ҫіНЁіЈІ»КЬҝШЈ¬ҝЙДЬ»бөјЦВІъЖ··ЗФӨЖЪ№ӨЧчЈ¬К№УГ·зПХПа¶ФҪПёЯЎЈ
ұн2іхКјКВјюәН»·ҫіКҫАэ
| НЁУГАаұр | іхКјКВјюәН»·ҫіКҫАэ | |
|---|---|---|
| І»НкХыөДТӘЗу |
РФДЬІ»·ыәПТӘЗуЈ» ЛөГчКйОҙ¶ФЙиұёј°ёҪјюО¬»ӨұЈСшөД·ҪКҪЎў·Ҫ·ЁЎўЖөҙОҪшРРЛөГчЈ» |
|
| ЦЖФм№эіМ |
ҝШЦЖіМРтј°ЙъІъ№ӨТХЎўЧчТөЦёөјКйРЮёДОҙҫӯСйЦӨЈ¬өјЦВІъЖ·ЦКБҝІ»ОИ¶ЁЈ» ЙъІъ№эіМ№Шјь№ӨРтҝШЦЖөгОҙҪшРРјаІвЈ¬өјЦВІъЖ·І»·ыәПТӘЗуөИЈ» Нв№әЎўНвРӯјю№©·ҪСЎФсІ»өұЈ¬Нв№әЎўНвРӯјюОҙҪшРРУРР§Ҫш»хјмСйЈ¬өјЦВІ»әПёсНв№әЎўНвРӯјюН¶ИлЙъІъөИЎЈ |
|
| ФЛКдәНЦьІШ |
ІъЖ··А»ӨІ»өұөјЦВЙиұёФЛКд№эіМЦРЛр»өөИЈ» ФЪі¬іцЙиұё№ж¶ЁөДЦьІШ»·ҫіЈЁОВ¶ИЎўКӘ¶ИЎўС№БҰЈ©ЦьІШЙиұёЈ¬өјЦВЙиұёІ»ДЬХэіЈ№ӨЧчөИЎЈ |
|
| »·ҫіТтЛШ |
ОВ¶ИЎўКӘ¶ИЎўәЈ°ОИзі¬іцёш¶Ё·¶О§әуҝЙДЬФміЙФЛРРІ»ХэіЈЈ» №эИИЎў№эАдөД»·ҫіҝЙДЬөјЦВЙиұёІ»ДЬХэіЈ№ӨЧчөИЈ» ЗҝЛбЗҝјоөјЦВЙЛәҰөИЈ» ҝ№өзҙЕёЙИЕДЬБҰІоЈ¬МШ¶Ё»·ҫіЙиұё№ӨЧчІ»ХэіЈөИЈ» ЙиұёөД№©өзөзС№І»ОИ¶ЁЈ¬өјЦВЙиұёІ»ДЬХэіЈ№ӨЧч»тЛр»өөИЎЈ |
|
| ІБКГәНЗеҪа | К№УГХЯОҙ°ҙТӘЗуҪшРРО¬»ӨЎўІБКГәНЗеҪаЎЈ | |
| ҙҰЦГәН·ПЖъ | ОҙФЪК№УГЛөГчКйЦР¶ФөНЖөөзБЖТЗ»тЖдЛыІҝјюөДҙҰЦГЈЁМШұрКЗК№УГәуөДҙҰЦГЈ©әН·ПЖъ·Ҫ·ЁҪшРРЛөГчЈ¬»тРЕПўІ»ід·ЦЈ»Оҙ¶ФЙиұё·ПЖъөДҙҰЦГҪшРРМбКҫРФЛөГчөИЎЈ | |
| ИЛОӘТтЛШ |
ЙијЖИұПЭТэ·ўөДК№УГҙнОуЈ» ЙијЖұдёьОҙУРР§ЦҙРРЈ» ТЧ»мПэөД»тИұЙЩК№УГЛөГчКйЈә —НјКҫ·ыәЕЛөГчІ»№ж·¶ —ІЩЧчК№УГ·Ҫ·ЁІ»Зеію —јјКхЛөГчІ»Зеію —ЦШТӘөДҫҜёжРФЛөГч»тЧўТвКВПоІ»ГчИ· —І»ККөұөДІЩЧчЛөГчөИ |
|
| К§Р§ДЈКҪ | УЙУЪАП»ҜЎўДҘЛр¶шөјЦВ№ҰДЬНЛ»Ҝ/ЖЈАНК§Р§ЎЈ | |
| ОЈПХЈЁФҙЈ© | ҝЙФӨјыөДКВјюРтБР | ОЈПХЗйҝц | ЙЛәҰ |
|---|---|---|---|
| өзҙЕДЬБҝ | ФЪЗҝөзҙЕ·шЙдФҙёҪҪьК№УГөНЖөөзБЖТЗІвБҝ | өзҙЕёЙИЕіМРтФЛРР | КЬөҪУ°ПмОЮ·ЁХэіЈФЛРР |
| ҫІөз·Еөз | ёЙИЕіМРтФЛРР | КЬөҪУ°ПмОЮ·ЁХэіЈФЛРР | |
| »јХЯФЪЙиЦГөзҙМјӨІОКэКұЈ¬Ҫ«ІОКэЙиЦГ№эҙу | »јХЯК№УГёГ№эҙуөДІОКэҪшРРҙМјӨ | ТэЖрИЛМеЧйЦҜҙМјӨІҝО»әмЦЧЎўЧЖЙЛ | |
| өзҙМјӨКдіцЛ«ПтХэёәВціеДЬБҝІ»ЖҪәв | »јХЯК№УГБЛөҘПтВціе»тІ»ЖҪәвЛ«ПтІЁҪшРРЦОБЖ | ТэЖрИЛМеЧйЦҜҙМјӨІҝО»өз»ҜС§·ҙУҰЈ¬ФміЙЛрЙЛ | |
| ИИДЬ | ·ЗФӨЖЪөД»т№эБҝөДөзј«Ж¬ЙэОВ | өзБчКдіц№ҰВК№эёЯ | ТэЖрИЛМеЧйЦҜ№эИИ»төјЦВММЙЛ |
| »ъРөДЬ | №ӨЧчЧҙМ¬ЦРТЖ¶ҜІъЖ·»тІЩЧчІ»өұ | ІъЖ·ҙУёЯҙҰЧ№Вд | ФТЙЛ»јХЯ»тІЩЧчХЯЈ»ЙиұёЛр»өөјЦВОЮ·ЁХэіЈК№УГ |
| ЙъОпС§ | К№УГУРЙъОпПаИЭРФІ»БјөДІДЦКЦЖЧч | ИЛМеҪУҙҘ | ЖӨ·ф№эГфЎўҙМјӨ |
| »ҜС§ | іӨКұјдІ»К№УГөДөзіШОҙҫӯИЎіцЈ¬ФміЙөзіШВ©Тә | өзВ·ёҜКҙ | Йиұё№КХПЈ¬ОЮ·Ё№ӨЧч |
| »·ҫі | ЙиұёКЬөҪНвҪзөДөзҙЕёЙИЕ | ІъЖ·ЙијЖКұөзҙЕЖБұОј°өзВ·ҝ№ИЕЙијЖІ»ід·Ц | І»ДЬХэіЈ№ӨЧч |
| Оҙ№ж¶ЁЙиұёөДК№УГ»·ҫі | |||
| Йиұё¶ФНвҪзөДөзҙЕ·шЙдёЙИЕ | ЖБұОЎўВЛІЁј°ҪУөШјјКхІ»НкЙЖ | ТэЖрЖдЛыЙиұёІ»ДЬХэіЈ№ӨЧч | |
| Оҙ№ж¶ЁЙиұёөДК№УГ»·ҫіТӘЗу | |||
| ЙиұёДЪІҝРЕәЕПЯУлөзФҙПЯПа»ҘёЙИЕ | |||
| ЖчРөК№УГ | К№УГХЯөДІЩЧчУРОуЎўОҙ°ҙЛөГчКйТӘЗуІЩЧч | ЙиұёК№УГІ»ХэіЈ | ҙпІ»өҪФӨЖЪЦОБЖР§№ы |
| УлПыәДЖ·ЎўёҪјюЎўЖдЛыТҪБЖЖчРөөДІ»ПаИЭРФ | ЕдМЧУГөДөзј«Ж¬І»ККУГЈ¬өјБӘПЯҪУҙҘ»төјНЁІ»Бј | ЙиұёОЮ·ЁХэіЈК№УГ | |
| Ҫ»ІжёРИҫ | УлК№УГХЯҪУҙҘөДІҝ·ЦЗеҪа/Пы¶ҫІ»ід·Ц»тІ»ХэИ· | ҝЙөјЦВёРИҫРФјІІЎ | |
| І»НкХыөДЛөГчКй | Оҙ¶ФҙнОуІЩЧчҪшРРЛөГч | ҙнОуІЩЧч | ҙпІ»өҪФӨЖЪЦОБЖР§№ыЈ¬СПЦШКұСУОуЦОБЖ |
| І»ХэИ·өДПы¶ҫ·Ҫ·Ё | К№УГУРёҜКҙРФөДЗеҪајБЎўПы¶ҫјБ | ІъЖ·ІҝјюёҜКҙЎў·А»ӨРФДЬҪөөН | |
| І»ХэИ·өДІъЖ·ЦьҙжМхјю | ЖчјюАП»ҜЎўІҝјюКЩГьҪөөН | ІъЖ·КЩГьҪөөН | |
| ҝЙДЬРиТӘёь»»өДБгІҝјюГ»УР№жёсЛөГч | К№УГІ»·ыәПТӘЗуөДЖчјю | ІъЖ·өДЛр»өЎўФміЙ°ІИ«Тю»јЈЁөзЖш°ІИ«Ј© | |
| ОҙЛөГчЛщРиөДО¬»Ө·Ҫ·Ё | І»КЗККөұөДО¬»Ө | ІъЖ·КЩГьҪөөН |
ЎЎЎЎұн2Ўўұн3ТАҫЭYY/T 0316өДёҪВјE МбКҫРФБРҫЩБЛІъЖ·ҝЙДЬҙжФЪОЈПХ(Фҙ)өДіхКјКВјюәН»·ҫіЈ¬КҫАэРФөШёшіцБЛОЈПХ(Фҙ)ЎўҝЙФӨјыөДКВјюРтБРЎўОЈПХЗйҝцәНҝЙ·ўЙъөДЙЛәҰЦ®јдөД№ШПөЈ¬ёшЙуІйИЛФұУиТФМбКҫЎўІОҝјЎЈ
ЎЎЎЎТҪБЖЖчРөЧўІбҙъАнУЙУЪөНЖөөзБЖТЗөДФӯАнЎў№ҰДЬәНҪб№№өДІоТмЈ¬ұҫХВёшіцөД·зПХТӘЛШј°ЖдКҫАэКЗіЈјыөД¶шІ»КЗИ«ІҝөДЎЈЙПКцІҝ·ЦЦ»КЗ·зПХ№ЬАн№эіМөДЧйіЙІҝ·ЦЈ¬І»КЗ·зПХ№ЬАнөДИ«ІҝЎЈЧўІбЙкЗлИЛУҰ°ҙХХYY/T 0316-2016Ў¶ТҪБЖЖчРө·зПХ№ЬАн¶ФТҪБЖЖчРөөДУҰУГЎ·ЦР№ж¶ЁөД№эіМәН·Ҫ·ЁЈ¬ФЪІъЖ·ХыёцЙъГьЦЬЖЪДЪҪЁБўЎўРОіЙОДјюәНұЈіЦТ»ёціЦРшөД№эіМЈ¬УГТФЕР¶ЁУлТҪБЖЖчРөУР№ШөДОЈПХ(Фҙ)Ўў№АјЖәНЖАјЫПа№ШөД·зПХЎўҝШЦЖХвР©·зПХІўјаКУЙПКцҝШЦЖөДУРР§РФЈ¬ТФід·ЦұЈЦӨІъЖ·өД°ІИ«әНУРР§ЎЈ
әиФ¶ТҪБЖЖчРөЧЙСҜ·юОс№«Лҫ КЗТ»јТјјКхЧЁТөөДТҪБЖЖчРөЧЙСҜ·юОс№«ЛҫЈ¬ЧЁЧўМṩȫ№ъёчөШИзЈәЙоЫЪЎў№гЦЭЎў¶«ЭёЎўЦРЙҪЎў·рЙҪЎўіұЦЭЎўЛіөВЎўЙПәЈЎўОч°ІЎўЦШЗмЎўіЙ¶јЎў°І»ХЎўҪӯЛХЎўХгҪӯөИЦӘГыіЗКРөДТҪБЖЖчРөБмУтјјКхЧЙСҜ·юОсЎЈәиФ¶ТҪБЖЖчРөЧЙСҜЧЁТө·юОсУЪЈәТҪБЖЖчРөІъЖ·ЧўІбЦӨҙъ°мАнЧЙСҜЎўТҪБЖЖчРөІъЖ··ЦАаҪз¶Ёҙъ°мАн,ҙъ°мТҪБЖЖчРөЙъІъРнҝЙЦӨЎўТ»АаТҪБЖЖчРөІъЖ·ұё°ёҙъ°мЎўМеНвХп¶ПКФјБЧўІбЎўҪшҝЪТҪБЖЖчРөЧўІбЎўТҪБЖЖчРөҫӯУӘРнҝЙЦӨҙъ°мЎў¶юАаТҪБЖЖчРөҫӯУӘұё°ёЎўCEИПЦӨЎўISO13485ИПЦӨЎўFDAЧўІбЎўFDAИПЦӨЎўБЩҙІКФСйЎўТҪБЖЖчРөЦКБҝ№ЬАнМеПөИПЦӨј°МеПөҪЁБўУл№эіМИ·ИПОДјюҪЁБў( ISO13485, GMP, CEЈ¬QSR820Ј¬CMDCAS);ЧўІбЎўТҪБЖЖчРөіцҝЪПъКЫЦӨГчЎўІъЖ·јјКхТӘЗуЦЖ¶©ЎўјјКхОДјюЎўН¬АаІъЖ·¶ФұИГвБЩҙІЖАјЫЧКБПұаРҙЎўЧўІбЧКБПұаРҙёЁөјЎўТҪБЖЖчРө№гёжЕъОДЙкұЁЎўөзҙЕјжИЭХыёДөИМṩһХҫКҪ·юОс№«ЛҫЈ¬»¶УӯДъЧЙСҜУләПЧчЈЎ
 ФЪПЯҝН·ю
ФЪПЯҝН·ю