

医疗器械注册从国际和国内情况来看,药械组合产品申报日益增多。由于药械结合产品的多样性和复杂性,申报注册中存在的问题也日益凸显。本文对药械组合产品属性界定情况进行介绍,汇总2009~2019年产品界定结果,并分析产品属性界定中的常见问题,希望为相关产品的研发和生产单位提供参考借鉴。
一、药械组合产品属性界定情况简介
目前,根据国家药品监督管理局(以下简称“国家局”)2009年11月发布的《关于药械组合产品注册有关事宜的通告》(2009年第16号)[1],拟申报注册的药械组合产品,如该类产品尚未在中国获准上市,申请人应当在申报注册前,向国家局行政事项受理服务和投诉举报中心(以下简称“受理中心”)申请产品属性界定。
国家局2009年建立了药械组合产品界定机制,受理中心收到申请人的属性界定申请后,组织相关单位进行界定,并将界定结果以书面通知的方式告知申请人;2015年制订了《药械组合产品属性界定工作程序》;2016年,将原有操作程序中由受理中心牵头,函询药品化妆品注册管理司、医疗器械注册管理司和药品审评中心(以下简称“药审中心”)、医疗器械技术审评中心(以下简称“器审中心”)四方意见,若各方意见存在分歧,受理中心组织各方召开产品属性界定会议投票决定产品属性的界定机制,简化为受理中心牵头,函询药审中心和器审中心,当两个审评中心意见不一致时,直接进入专家会议决定的流程。药械组合产品属性界定程序的改进,减少了中间环节,缩短了出具界定结果所需的时间周期,提高了界定的效率。2019年,药械组合产品属性界定工作改由局医疗器械标准管理研究所(医疗器械标准管理中心)承担。
在我国,药械组合产品系指由药品与医疗器械共同组成,并作为一个单一实体生产的产品。产品属性的界定是依据药械组合产品的定义和产品的主要作用方式,来进行判定的,并以不造成药品、医疗器械管理交叉为基本原则。
以药品作用为主的药械组合产品,申报药品注册,由药审中心牵头审评;以医疗器械作用为主的药械组合产品(以下简称“含药器械”),申报医疗器械产品注册,由器审中心牵头审评。需要联合审评的药械组合产品,注册申报资料中相应部分由牵头审评中心转交协作审评中心同步进行审评。鉴于药品和医疗器械在质量体系、检测、申报要求和上市后监管等各方面存在较大差异,为减轻申请人负担,鼓励申请人在该类产品研发早期阶段进行沟通咨询和产品属性界定,避免出现后期的注册路径和申报资料准备的不合规。
二、药械组合产品属性界定结果汇总
《关于药械组合产品注册有关事宜的通告》中,已明确“带药物涂层的支架、带抗菌涂层的导管、含药避孕套、含药节育环等产品,按医疗器械进行注册管理,含抗菌、消炎药品的创口贴、中药外用贴敷类产品等按药品进行注册管理”。但由于药械组合产品的形式多样,不同国家或地区对组合产品的定义和监管模式不一致,以及部分产品本身的主要作用方式尚不明确或存在争议等原因,导致某些产品在不同国家或地区的监管方式存在差异,每年仍有相当数量的产品申请进行属性界定。
目前,常见申请属性界定的产品包括含药品涂层或药品浸渍或与药品结合的器械、预填充药品给药装置/系统、预填充生物制品给药装置/系统、生物制品涂层或结合生物制品的器械、外用液体或凝胶产品等。
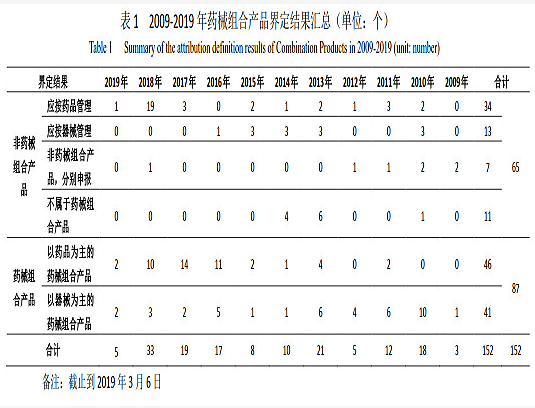
国家局为提高药械组合产品界定结果的透明度,引导申请人合理申报,公布了历次产品属性界定的结果。截至2019年3月6日,国家局已发布药械组合产品属性界定结果公告12次,累计完成产品属性界定152个,其中被界定为药械组合产品的87个,占总量的57.2%,非药械组合产品的65个,占总量的42.8%;被界定为药械组合产品的87个产品中,以药品作用为主的药械组合产品46个,占总量的52.9%;以医疗器械产品作用为主的药械组合产品41个,占总量的47.1%。
三、药械组合产品属性界定资料中的常见问题分析
医疗器械注册咨询据国家局器审中心联合药审中心、中国食品药品检定研究院和受理中心等多家单位共同承担了“药械组合产品有关管理政策研究”课题,明确提出了药械组合产品属性界定申请资料清单,在多次对外交流培训中进行宣讲,但申请人提交的属性界定申报资料中仍存在以下几个常见问题:
1.提交的产品基本信息不完整
建议申请人在属性界定申请资料中明确产品的基本信息。对于首次进口的药械组合产品,建议明确产品是否获出口国(地区)批准上市,申请资料中建议明确产品的组成及各组件(分)的用途、产品示意图等,拟采用的产品使用说明书及标签;对于组合产品中的药品部分,建议明确药品的名称、供应商、是否具有在我国注册或生产国(地区)批准上市的证明文件,在组合产品中使用时与其作为药品单独上市时在预期用途/适应证、接触途径、剂量、禁忌症等方面存在的差异等;对于组合产品中的器械部分,建议明确器械的结构组成,若单独已作为医疗器械上市,建议提交上市证明文件,以及在组合产品中使用时与其作为医疗器械单独上市时在预期用途/适用范围、性能要求等方面存在的差异等。
2.药械组合产品的立题依据不充分
医疗器械注册代办建议注册申请人在研发早期充分考虑药械组合产品的立题依据。以含药器械为例,在医疗器械中的添加使用药物等活性成分,建议论证添加活性成分的立题依据是否合理,不鼓励在器械上盲目添加活性成分。例如,在医疗器械中添加抗菌成分,带来收益的同时也存在风险,抗菌成分可随器械直接与局部组织接触,甚至突破人体的血药屏障,改变抗菌成分原有暴露途径,建议研发时考虑所用抗菌成分是否有相应的临床使用史和耐药性等问题,同时需考虑其是否能实现预期的局部抑菌效果。
含药器械的研发、生产企业多为医疗器械科研机构或制造企业,常缺乏药学相关专业的技术人员,在开发药械组合产品时应更为慎重。
3.产品作用机理及主要作用方式缺乏支持资料
医疗器械注册代理建议申请人在属性界定申请资料中提交支持资料论证产品的作用机理及主要作用方式。部分申请人在申请资料中提出产品中所含成分不具有活性,但未能提交充分的支持资料予以证明所述观点。2013年,国家局部署开展贴敷类医疗器械注册专项检查工作[2],明确含有化学成分、中药材(或天然植物)及其提取物的贴敷类产品,无论药典是否收载,或者是否属于药食同源,如所添加成分发挥药理作用的,不能按照医疗器械审批。
4.未明确产品中药品部分和器械部分的组合方式及产品的使用方法或步骤
医疗器械注册办理建议申请人在属性界定申请资料中明确产品中药品部分和器械部分的组合方式,以及产品的使用方法或使用步骤,以便判断产品是否符合药械组合产品的定义。例如,申请界定的含抗凝剂血液成分分离器械,根据其说明书中的产品使用方法,该产品中抗凝剂由药品公司生产和供应,是个单独的灭菌包装,与血液成分分离器械打包放在一个套装内,在使用时用注射器抽取抗凝剂预充至分离器械。上述情形不符合我国药械组合产品的定义,因此该产品界定结果为不属于药械组合产品,应分别申报。
四、展望
随着科学技术的发展,更多创新型组合产品不断刷新现有药械组合产品的概念,数字化药物、组织工程医疗产品、药械“融合”产品等新型组合产品纷纷涌现,例如美国FDA在2017年11月批准的全球首个数字化药物[3],由含传感器的智能口服药丸、智能贴片、智能设备App、基于Web的数据面板4部分组成。
面对创新型组合产品不断涌现的大趋势,监管机构需紧密跟踪国内外组合产品发展前沿及监管经验,建立适合我国的组合产品科学界定原则,以备在创新型组合产品研发成熟度不断提高的情况下,能够科学引导、合理监管。
深圳鸿远医疗器械咨询服务公司 是一家技术专业的医疗器械咨询服务公司,专注提供全国各地如:深圳、广州、东莞、中山、佛山、潮州、顺德、上海、西安、重庆、成都、安徽、江苏、浙江等知名城市的医疗器械领域技术咨询服务。鸿远医疗器械咨询专业服务于:医疗器械产品注册证代办理咨询、代办医疗器械生产许可证、一类医疗器械产品备案代办、医疗器械经营许可证代办、二类医疗器械经营备案、进口医疗器械注册、CE认证、ISO13485认证、FDA注册、FDA认证、临床试验、医疗器械质量管理体系认证及体系建立与过程确认文件建立(ISO9001, ISO13485, GMP, CE,QSR820,CMDCAS);注册、出口证、自由销售证办理、产品技术要求制订、技术文件、临床试验及免临床同类产品对比资料编写、注册资料编写辅导、电磁兼容整改、医疗器械广告批文申报等提供一站式服务公司,欢迎您咨询与合作!
 在线客服
在线客服