

��
�ڶ���ҽ����е��Ʒע��֤�������ݡ�ҽ����е�ල��������������ҽ����еע������취����ҽ����еע��֤������Ч��Ϊ���꣬��������Ҫ��������ȡ�õġ�ҽ����еע��֤������Ч�ڵģ�Ӧ������Ч�ڽ���ǰ6����������롣
����ҽ����еע��֤������������
��������ҽ����еע��֤�������������ˣ�Ӧ��������������
����(һ)ԭҽ����еע��֤Ϊ�㶫ʡʳƷҩƷ�ල�����ֺ˷�������ʱ���յڶ���ҽ����е������(����ԭע��֤Ϊ�����ֺܾ˷�������ҽ����е֤������ʱ��Ʒ������Ϊ�����ҽ����е)��
����(��)ע��֤��Ч�ڽ���6����ǰ��������ע�ᡣ
����(��)�������Ϸ���Ҫ��
����ҽ����е��Ʒע��֤�����������
����һ���������Ŀ¼
����(һ)�����
����(��)֤�����ļ�
����(��)���ڲ�Ʒû�б仯������
����(��)ԭҽ����еע��֤��������ӡ������Ӧ������ҽ����еע�����ļ���ӡ����
����(��)ע��֤��Ч���ڲ�Ʒ������������
����(��)��Ʒ���鱨��
����(��)����������
����(��)����
����(��)�����ļ�
�����������������ʽ��
����(һ)�������Ӧ���������࣬ʹ��A4���ֽ�Ŵ�ӡ��
����(��)�걨����Ӧ��дĿ¼���걨����ÿ���ļ���һҳ����ǩ�����ô���ǩ�ĸ�ҳֽ�ָ��������걨����Ŀ¼����ÿ���ļ���š�ȫ���걨���ϰ���Ŀ¼˳��װ���ɲᡣ
����(��)ÿ���ļ���Ӧ�Ӹ���ҵ����;
����(��)�걨����һʽһ�ݣ�(�����һʽ���ݣ�һ�����걨����װ��һ����һ�ݲ���Ҫװ��)��Ӧ��ʹ��A4���ֽ�Ŵ�ӡ���������������������Ϳ�ġ���װ���ɲ�ģ��������в�֡�
����(��)�걨����ʹ�ø�ӡ���ģ���ӡ��Ӧ����������ԭ��һ�¡��걨������ע���ύԭ���ģ����밴��Ҫ���ύԭ����
����(��)�����걨�����е���������Ӧ������һ���ԡ�
����(��)�걨���Ͼ�Ӧ�Ӹ������˹��¡�
����(��)ҽ����еע���걨���ϻ���ͬʱ�ύ���µ����ļ�:(a)�������(b)��Ʒ����Ҫ��ӦΪword�ĵ������ҿɱ༭���ġ�ͬʱ��Ӧ�ύ�����Ľ���������Ҫ������ָ�겿�ֵĵ����ĵ���ӦΪword�ĵ���
��������ҽ����еע��֤�����걨���ϵľ���Ҫ��
����(һ)�����
�������˵�����£�
����1.����Ӧͨ����ҵ���ϰ���ƽ̨�����걨�˿ڵ�½������������ߴ�ӡ���Ӹ���ҵ���¡�
����2.�����������Ӹ���ҵ���£����Ӹ�����¡������“��֤��”��Ŀ�е�������ǩ�£���ָ���������˻�����ǩ�����Ӹ���ҵӡ�¡�
����3.Ҫ����д����Ŀ����ԭ����ʹ�����ġ���д���������������ÿհף���������ݴ�Ӧ��д“�M”�����������ʽ��������д����ʱ��������������
����4.��ֽ���걨�ģ��ύԭ��ɨ���pdf�ĵ�������ֽ���걨�ģ��ύԭ����
����5.ǰ��ҽ����е��Ʒע���������ϵָ�ò�Ʒ���뱾��ע���������һ��δ��ע��������
����6.�ͺš����Ӧ�����ύ�������ʵ�����������Ӧ��
����7.��������Ӧ�ṩ��ҽ����е����Ŀ¼���ľ���Ʒ��(��Ӧ��Ʒ������)�����ṩ����綨֪ͨ�ļ��еľ�����������綨���֪ͨ�顣
����8.��Ʒ��������Ӧ���ݡ�ҽ����е����Ŀ¼�������綨���������ļ���д��
����9.������ס������д������Ӫҵִ�յ����֤�����ļ���������ס����
����10.���������ڵ�ϵָ������ס�������С�
����11.������ַ��ָ��Ʒʵ�ʼӹ�����ĵ�ַ��
����12.����������Ҫ�ر����˵�������⣬���ڱ���“������Ҫ˵��������”����˵����
����(��)֤�����ļ�
����ҽ����еע��֤����ע�����ṩ��ҵӪҵִ�յĸ�����ӡ������֯��������֤��ӡ��;
����˵����Ӫҵִ��Ҫ��Ϊ������ӡ������ӡ���������Ӹ���ҵ���¡�
����(��)���ڲ�Ʒû�б仯������
����ҽ����е��Ʒע��֤����ע�����ṩ��Ʒû�б仯������(ע��֤�������ļ������������ݡ���Ʒ���漰����ع��ұ�����ҵ��û������û���µ���ع��ұ�����ҵ��������Ӱ���Ʒ��ȫ��Ч����ơ�ԭ���ϡ��������ա����÷�Χ��ʹ�÷�����)��
����(��)ԭҽ����еע��֤��������ӡ������Ӧ������ҽ����еע�����ļ���ӡ����
����(��)ע��֤��Ч���ڲ�Ʒ������������
����1����Ʒ�ٴ�Ӧ��������û�Ͷ���������ȡ�Ĵ�ʩ��
����2��ҽ����е�����¼����ܷ������۱��棬����Ӧ�Ա���Ʒ���к����Ŀ��ɲ����¼��б���˵����ÿһ�������������ҵ��ȡ�Ĵ����ͽ�������������������¼����з������ۣ����������¼�������ԭ���䰲ȫ�ԡ���Ч�Ե�Ӱ������˵����
����3�������й��Һ͵����IJ�Ʒ�г����˵����
����4����Ʒ�ල�������(����)��
����5�������к������ٻأ�Ӧ˵���ٻ�ԭ���̺ʹ��������
����6��ԭҽ����е��Ʒע��֤������Ҫ�������ɹ����ģ�Ӧ���ṩ����ܽᱨ�棬������Ӧ���ϡ�
����7����Ʒ���鱨��
��������ص�ҽ����еǿ���Ա��Ѿ�����Ӧ�ṩ��Ʒ�ܹ��ﵽ��Ҫ��IJ�Ʒ���鱨�档��Ʒ���鱨��������Լ챨�桢ί�м��鱨������ǿ���Ա�ʵʩ֪ͨ�涨�ļ��鱨�档���У�ί�м��鱨��Ӧ�ɾ���ҽ����е�������ʵ�ҽ����е����������ߡ�
������Ϊ����ǿ���Ա�Ҫ���Ʒ���������ģ������漰��Ʒ���ܰ�ȫ�ĸı�Ҫ�����漰�������ܰ�ȫ���м�⣬�������Ҫ����ͬһ��������������ⱨ�棬���ݼ�ⱨ��Ҫ���й�����
����(��)����������
����1��ҽ����еע������������Ʒ���ϡ�ҽ����еע������취������ط����Ҫ��;��������Ʒ�������й��ұ�����ҵ�������ṩ���ϱ����嵥;
����2��ע���˳������ύ������ʵ�Ե����ұ�֤������
����(8)����
����1������ԭҽ����еע��֤��Ч���ڷ������漰��Ʒ����Ҫ�����ģ�Ӧ���ύ����ע�����ļ��ĵIJ�Ʒ����Ҫ��һʽ���ݣ����ύ���ݲ�Ʒ����Ҫ���ı���ȫһ�µ�������
����2��2014��10��1��ǰ�ѻ�ע��������ע��ʱ��ע���˰��ա�ҽ����еע������취���涨�ύ���ϣ�ͬʱ�ύԭע���Ʒ��ԭ������Ʒ����Ҫ��Ʒ����Ҫ����ԭע���Ʒ���ĶԱ�˵��;��С���۵�Ԫ�ı�ǩ�������;��˵������ԭ��ע������˵�����б仯�ģ�Ӧ�ṩ��������Ա�˵����
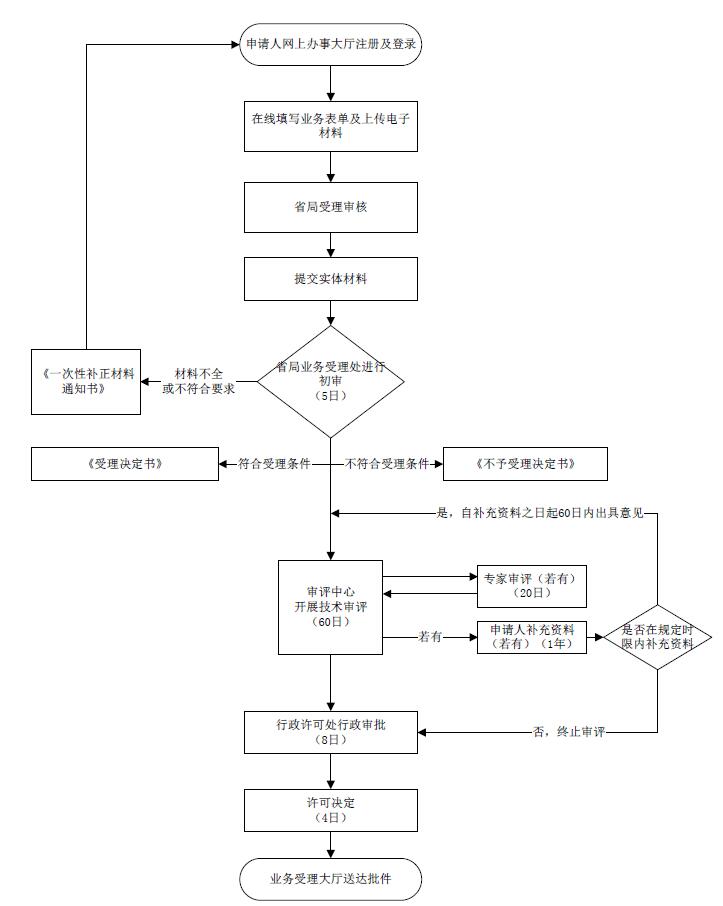
����ҽ����е��Ʒע��֤�����������̣�
�����ڶ���ҽ����еע��֤�����������ڰ������̣� �걨�˱���ͨ���Ҿֵ���ҵ���ϰ���ƽ̨����ʡ�������ϰ��´������������걨�����貹��ֽ�ʲ��ϣ����������걨��ɺ�5���������ڼĴ�ʡ��ҵ������������ִ�ʡ��ҵ�����������еݽ���ע���������������ɴ�������ʽ��飬���Ϻϸ�ת�����������Ľ��м���������������������Ҫ��ת�������ɴ���������������������֤��ᣬ��ʡ����������֪�����˲���֤��
���� ҽ����еע��֤�����������ϰ������� ���걨�˱���ͨ���Ҿֵ���ҵ���ϰ���ƽ̨����ʡ�������ϰ��´������������걨�����貹��ֽ�ʲ��ϣ����������걨��ɺ�5���������ڼĴ�ʡ��ҵ������������ִ�ʡ��ҵ�����������еݽ���ע���������������ɴ�������ʽ��飬���Ϻϸ�ת�����������Ľ��м���������������������Ҫ��ת�������ɴ���������������������֤��ᣬ��ʡ����������֪�����˲���֤��
���ں�Զҽ����е��ѯ��˾ http://www.yixiezixun.com��һ�Ҽ���רҵ��ҽ����е��ѯ������רע�ṩȫ�������磺���ڡ����ݡ���ݸ����ɽ����ɽ�����ݡ�˳�¡��Ϻ��������������֪�����е�ҽ����е��������ѯ����Զҽ����е��ѯרҵ�����ڣ�ҽ����е��Ʒע�������ѯ������ҽ����е��������֤��һ��ҽ����е��Ʒ�������졢ҽ����е��Ӫ����֤���졢����ҽ����е��Ӫ������CE��֤��ISO13485��֤��FDAע�ᡢFDA��֤���ٴ����顢ҽ����е����������ϵ��֤����ϵ���������ȷ���ļ�����(ISO9001, ISO13485, GMP, CE��QSR820��CMDCAS);ע�ᡢ����֤����������֤��������Ʒ����Ҫ���ƶ��������ļ����ٴ����鼰���ٴ�ͬ���Ʒ�ȶԱ����ϱ�д��ע�����ϱ�д������ҽ����е��������걨���ṩһվʽ�ķ����������ӭ����ѯ�������
 ���߿ͷ�
���߿ͷ�