

第三类高风险医疗器械临床试验审批服务指南
一、适用范围
本指南适用于第三类高风险医疗器械临床试验审批的申请和办理
二、项目信息
(一)项目名称:第三类高风险医疗器械临床试验审批
(二)子项名称:无
(三)审批类别:行政许可
(四)项目编码:30018
三、办理依据
《医疗器械监督管理条例》(国务院令第650号)第十九条:第三类医疗器械进行临床试验对人体具有较高风险的,应当经国务院食品药品监督管理部门批准。临床试验对人体具有较高风险的第三类医疗器械目录由国务院食品药品监督管理部门制定、调整并公布。
四、受理机构
国家食品药品监督管理总局医疗器械技术审评中心
五、决定机构
国家食品药品监督管理总局
六、审批数量
无数量限制
七、办事条件
境内申请人应为境内依法进行登记的企业。境外申请人应为境外生产企业,且该医疗器械已在注册申请人注册地或者生产地址所在国家(地区)已获准上市销售。申请的医疗器械应列入《关于发布需进行临床试验审批的第三类医疗器械目录的通告》(2014年第14号)的目录中。
八、申请材料
(一)申请材料清单
医疗器械临床试验审批申报资料
1.申请表
2.证明性文件
(1)境内申请人应当提交:企业营业执照副本复印件;组织机构代码证复印件。
(2)境外申请人应当提交:境外申请人注册地或生产地址所在国家(地区)医疗器械主管部门出具的允许产品上市销售的证明文件和合法资格证明文件;境外申请人在中国境内指定代理人的委托书、代理人承诺书及营业执照副本复印件或者机构登记证明复印件。
3.试验产品描述
应当包括试验用医疗器械的设计原理、工作原理、产品特征、结构组成及图示、制造材料、包装材料、型号规格及其划分依据、主要生产工艺、交付状态、作用机理、适用范围等内容。
4.临床前研究资料
一般应当包括:
(1)申请人对试验用医疗器械进行的临床前研究资料。例如,实验室研究、动物试验等。
(2)与评价试验用医疗器械安全性和有效性相关的已发表文献及评论性综述。
(3)国内外同类产品研发、上市及临床应用情况及试验用医疗器械与国内外已上市同类产品在工作原理、结构组成、制造材料、技术参数及适用范围等方面的异同比较资料。
(4)与试验用医疗器械相关的不良事件信息。
(5)临床试验受益与风险对比分析报告。
(6)其他要求提交的研究资料。
5.产品技术要求
6.医疗器械检验机构出具的医疗器械注册检验报告和预评价意见
7.说明书及标签样稿
8.临床试验方案
临床试验方案应当符合国家食品药品监督管理总局发布的《医疗器械临床试验质量管理规范》相关要求,并提交证明临床试验方案科学合理性的分析资料。
9.伦理委员会同意临床试验开展的书面意见
应当提交全部临床试验机构的伦理委员会同意临床试验开展的书面意见。
10.符合性声明
(1)申请人声明本产品符合《医疗器械注册管理办法》和相关法规的要求。
(2)申请人声明所提交资料的真实性。
(二)申请材料提交
申请人可通过窗口报送、邮寄等方式提交材料。
九、申请接收
(一)接收方式
1.窗口接收;
2.邮寄接收。
接收部门:国家食品药品监督管理总局行政受理服务大厅
接收地址:北京市西城区宣武门西大街28号大成广场2门一层
邮政编码:100053
联系电话:010-88330000
(二)对外办公时间:上午:9:00—11:30 下午:13:00—16:00
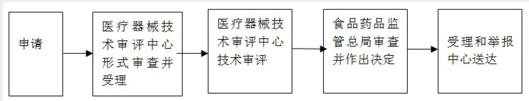
十、办理基本流程
十一、办理方式
1.受理:
申请人按照本《指南》第八条要求,向国家食品药品监督管理总局行政受理服务大厅提出申请,受理人员按照《关于公布医疗器械注册申报资料要求和批准证明文件格式的公告》(2014年第43号)的要求对申报资料进行形式审查。
申请事项属于本部门职权范围,申报资料齐全、符合形式审查要求的,予以受理;申报资料存在可以当场更正的错误的,允许申请人当场更正;申报资料不齐全或者不符合形式审查要求的,在5个工作日内一次告知申请人需要补正的全部内容,逾期不告知的,自收到申报资料之日起即为受理;申请事项不属于本部门职权范围的,即时告知申请人不予受理。
2.审查:
受理人员自受理之日起3个工作日内将申报资料转交技术审评机构。
技术审评机构应当在40个工作日内完成技术审评工作。
3.许可决定:
国家食品药品监督管理总局应当在技术审评结束后20个工作日内作出决定。准予开展临床试验的,发给医疗器械临床试验批件;不予批准的,应当书面说明理由。
4.送达:
自作出审批决定之日起10个工作日内,总局行政事项受理服务和投诉举报中心将行政许可决定送达申请人。
十二、审批时限
1.受理:5个工作日;
2.行政许可决定:20个工作日(不含技术审评和申请人补充资料及补充资料审评所需的时间)。20个工作日内不能做出决定的,经总局领导批准,可延长10个工作日。
十三、审批收费依据及标准
(一)收费环节:受理
(二)收费项目:第三类高风险医疗器械临床试验审批
(三)收费依据:国家发展改革委《关于重新发布中央管理的食品药品监督管理部门行政事业性收费项目的通知》(财税〔2015〕2号)和《关于印发〈药品、医疗器械产品注册收费标准管理办法〉的通知》(发改价格〔2015〕1006号),《国家食品药品监督管理总局关于发布药品、医疗器械产品注册收费标准的公告》(2015年第53号)。
(四)收费标准: 4.32万元。
十四、审批结果

十五、结果送达
自作出审批决定之日起10个工作日内,总局行政事项受理服务和投诉举报中心将行政许可决定送达申请人。
十六、申请人权利和义务
(一)依据《中华人民共和国行政许可法》,申请人依法享有以下权利:
1.依法取得行政许可的平等权利;
2.对行政机关实施行政许可,享有陈述权、申辩权;
3.依法申请行政复议或者提起行政诉讼;
4.合法权益因行政机关违法实施行政许可受到损害的,有权依法要求赔偿。
(二)依据《医疗器械注册管理办法》第三十六条,受理注册申请的食品药品监督管理部门对不予注册的,应当书面说明理由,并同时告知申请人享有申请复审和依法申请行政复议或者提起行政诉讼的权利。
(三)依据《中华人民共和国行政许可法》、《医疗器械注册管理办法》等,申请人应履行以下义务:
1.对申请材料实质内容的真实性负责;
2.依法开展取得行政许可的活动
3.如实向负责监督检查的行政机关提供有关情况和材料
鸿远医疗器械咨询 http://www.yixiezixun.com有限公司专业代办医疗器械产品注册证,医疗器械注册代理咨询,医疗器械生产许可证,医疗器械经营许可证,医疗器械生产备案,医疗器械经营备案,CE认证,FDA注册/认证,ISO13485认证,临床试验,医疗器械质量管理体系认证及体系建立与过程确认文件建立(ISO9001, ISO13485, GMP, CE,QSR820,CMDCAS);注册,出口证,自由销售证办理,临床试验及免临床资料编写,产品技术要求制订,技术文件编写辅导等提供一站式服务!
 在线客服
在线客服