

医疗器械FDA注册流程是什么? 医疗器械FDA注册有哪些要求?今天小编为大家整理了相关最新医疗器械FDA注册要求及流程如下:
一、明确FDA对医疗器械的分类
FDA将医疗用品分为三类,并采取不同的管理和控制:
(1)第一类医疗器械Class I:低等风险(监管控制类型:基本控制)
产品必须合乎一般规定要求,大部分可以直接注册,无需递交产品安全有效性报告;
(2)第二类医疗器械Class II:中等风险(监管控制类型:基本控制以及特殊控制)
产品必须达到功能标准;大部分需要注册前向FDA递交FDA510(K) (PMN市场预投放通告)的产品本身的安全有效性论证报告,获批后才可以进行产品注册和合法上市销售;申请周期在半年以上;510(K)仅对产品进行书面论证,不涉及获批前的工厂质量体系FDA GMP QSR820的现场审核;
(3)第三类医疗器械Class III:高等风险(监管控制类型:基本控制以及上市前批准)
最严格控制,上市前必须先经批准。大部分需要先申请FDA PMA市场预投放批准,获批后才可以进行产品注册和合法上市销售;申请周期在一年以上;获批前FDA会对制造商进行FDA GMP QSR820质量体系的现场审核,通过后才可获批该产品的PMA申请。
二、FDA对医疗器械的相关法规

美国法典CFR(CODE OF FEDAL REGULATION)的第21篇食品和药品中对医疗器械的通则、标签、分类、注册、豁免、召回等环节的标准、要求作出了详尽的规定。
三、FDA医疗器械注册程序
(一)选择正确的路径递交
器械分类确定之后,需要选择相应法规要求下的上市前递交。常见的上市前递交类型包括:
• 510(k)(上市前通知)
• PMA(上市前批准)
• De Novo(自动III类指定的评价)
• HDE (人道主义器械豁免)
Ⅰ类以及大部分Ⅱ类器械要求以510(k)的方式递交。在510(k)递交过程中,申请者必须证明新的器械与对比器械在预期用途,技术特征以及性能测试方面实质等同。一些Ⅰ类和Ⅱ类器械可以豁免510(k),如果他们在21 CFR 862-892.9所述的豁免范围之内。这些豁免被列在21 CFR的分类规则中,也被汇集在医疗器械豁免文件中。
大部分Ⅲ类器械要求的递交方式为PMA。PMA为严格程度较高的上市前递交类型。在FDA批准PMA之前,申请者必须提供有效的科学证据,以证明器械预期用途的安全性以及有效性。
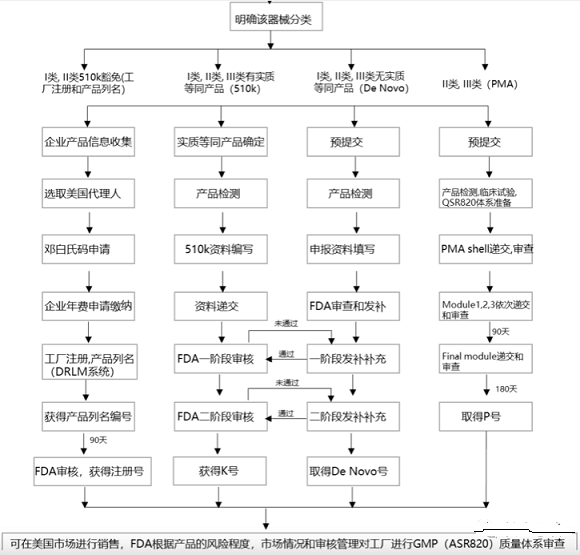
De Novo为没有有效对比的新器械提供一种方式,如果这种新器械满足特定标准,可以被分为Ⅰ或Ⅱ。
HDE为Ⅲ类器械提供了一种监管路径,这类器械预期对罕见疾病或状况的患者是有益的。器械有资格成为人道主义豁免器械,申请者必须获得人道主义使用器械(HUD)的指定,可通过向FDA的孤儿产品开发办公室Office of Orphan Products Development (OOPD)递交申请。
(二)准备材料
在选择正确的上市前递交类型之后,必须准备该递交类型所需的适当的资料。FDA开发一些帮助申请者准备上市前递交的资源类型,包括:
1.器械建议(Device Advice):—综合基于FDA上的网页法规协助
2.510(k)的准备:参考Premarket Notification 510(K)
3.PMA的准备:参考Premarket Approval (PMA)
4.CDRH学习(CDRH Learn):基于视频的教学模块,研讨会和录制的包括各种政策和指导力度的网络研讨会,CDRH递交前程序 —未来上市前提交申请可能要求FDA通过这个程序进行反馈。
准备上市前递交时需要考虑的信息:
(1)设计控制:所有II类以及III类器械根据质量管理体系(21 CFR 820.30)中对设计控制的要求进行设计。一些I类器械可豁免设计控制。
(2)非临床测试:器械上市要求的测试以及信息类型是通过器械的分类,作用机制,技术特征,以及标签来确定的。医疗器械上市前递交实施的非临床测试必须符合21 CFR 58中的良好试验管理规范(GLPs)
(3)临床证据:PMAs,HDEs 以及部分 510(k)s 和 De Novos 要求有临床证据。在早起的临床研究开始之前,研究申请者需要得到FDA器械临床研究豁免(IDE)的批准。这项研究也需要得到伦理审查委员会(IRB)的批准。临床研究必须符合所有的适用的器械临床研究豁免(IDE)法规以及良好试验管理规范(GLPs)。
(4)标签:器械的标签必须依据标签法规书写,且需要包含在上市前递交的资料中。
(三)递交资料
提交给FDA,并在FDA的工作人员审查过程中保持联系。
1.用户费用:在510(k)或PMA递交时,需要一定的用户费用
2.电子副本(eCopy):上市前递交必须包含以光盘(CD)、数字视频光盘(DVD),或闪存驱动器方式形成的电子副本。
3.行政备案审查:在上市前递交接收之后,FDA进行行政审查,评估递交是否是足够完整的,以接收实质性审查。
4.审查互动(Interactive Review):当递交的资料处于正在审查中时,FDA将和申请者保持联系以增加审查过程中的效率。
(四)完成登记
医疗器械FDA注册代办器械设备必须在FDA对其生产的企业进行登记,并对其器械进行列名。如果一个器械在上市前需要上市前清关(premarket clearance)或上市前批准(premarket approval),器械厂商在登记和列名之前必须等到它获得FDA的清关或批准。器械企业登记、登记号的分配或医疗器械的列名,都不意味着FDA对其企业或其产品的清关或批准。
 在线客服
在线客服